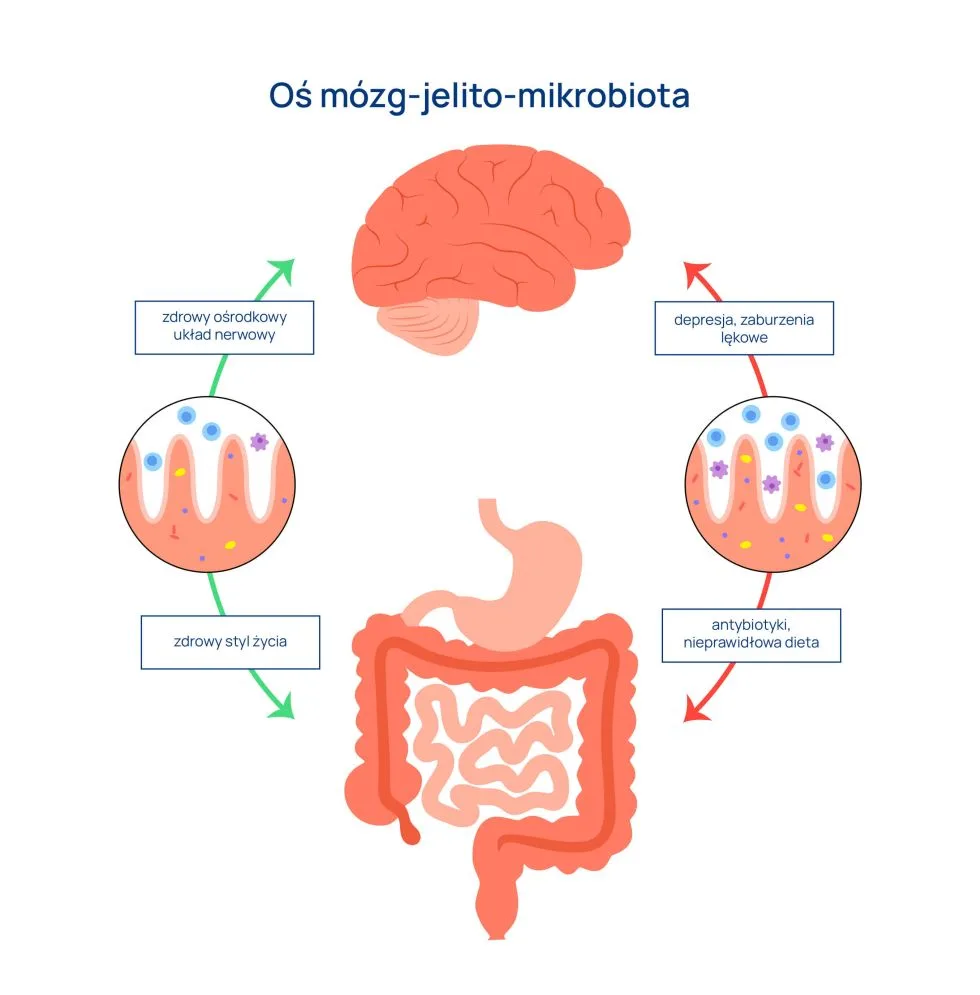
W 1980 roku dość niespodziewanie została zaprezentowana koncepcja osi jelitowo-mózgowej w wyniku przeprowadzonych badań nad zależnością pomiędzy hormonami a układem nerwowym (Track, 1980). Od tego czasu minęły cztery dekady intensywnych badań, które rozszerzyły koncepcję osi, nazywając ją osią mózg-jelito-mikrobiota. Okazało się bowiem, że bardzo ważny elementem, będącym źródłem wysyłanych sygnałów jest mikrobiota jelitowa.
Oś mózg – mikrobiota – jelito
Wiemy już, że przekaz informacji w zakresie osi mózg-jelito-mikrobiota jest dwukierunkowy i angażuje następujące drogi:
- Nerw błędny
- Hormony i neurotransmitery
- Szlaki neuroendokrynne: przede wszystkim oś podwzgórze-przysadka-nadnercza z kluczową rolą kortyzolu
- Szlaki układu odpornościowego
- Metabolity pochodzenia bakteryjnego: zwłaszcza krótkołańcuchowe kwasy tłuszczowe
- Czynniki neurotroficzne
Mikrobiota jelitowa składa się głównie z bakterii, wirusów, archeonów i grzybów. Liczebność bakterii w gramie treści pochodzącej z jelita grubego wynosi do 1012 komórek. To powoduje, że stosunek komórek bakteryjnych do ludzkich w organizmie wynosi prawie 1:1. Natomiast genom mikrobiomu jelitowego zawiera ponad 100-krotnie więcej genów niż genom ludzki. To powoduje, że nasze spojrzenie na jelita i zamieszkującą je mikrobiotę jelitową jako na nasz „drugi mózg” staje się zasadne i poparte obserwacjami naukowymi.
Pytaniem ciągle otwartym pozostaje zakres możliwego wpływu mikrobioty jelitowej na funkcjonowanie naszego ośrodkowego układu nerwowego. Pytanie to jest o tyle zasadne, że w ostatniej dekadzie pojawiły się wyniki prac badawczych, które jednoznacznie wskazują na udział wybranych gatunków i szczepów bakteryjnych pochodzących z jelit w kształtowaniu obrazu klinicznego chorób psychiatrycznych.
Badania wpływu mikrobioty jelitowej na depresję
W badaniach przeprowadzonych przez Peter i wsp. w grupie pacjentów z zespołem jelita nadwrażliwego pacjenci z lękiem charakteryzowali się podwyższoną liczebnością Bacteroidaceae (rodziny Bacteroidaceae i Rikenellaceae). Natomiast typ Proteobacteria i rodzina Prevotellaceae liczniej reprezentowane były u pacjentów z depresją. Bardzo ciekawą okazała się obserwacja odwrotnej korelacji liczebności Lachnospiraceae z nasileniem objawów depresyjnych.
Badania przeprowadzone w grupie polskich kobiet chorujących na depresję dostarczyły bardzo ciekawych wniosków. Okazuje się, że istnieją różnice w stężeniach metabolitów pochodzenia bakteryjnego w stolcu osób chorych na depresję w porównaniu do osób zdrowych. Kobiety bez depresji miały wyższe stężenia wszystkich krótkołańcuchowych kwasów tłuszczowych (SCFA), z wyjątkiem kwasu izokapronowego, w porównaniu z kobietami z depresją. Interesująca była też analiza pozwalająca stwierdzić, że stężenie kwasu octowego, propionowego i izokapronowego było odwrotnie proporcjonalne do nasilenia objawów depresyjnych.
Jedno z przeprowadzonych przez badaczy amerykańskich badań uwzględniających teorię wpływu stanu zapalnego i zjawiska translokacji wykazało, że u osób z bardziej wrogimi interakcjami małżeńskimi notowane były wyższe stężenia endotoksyny bakteryjnej we krwi w porównaniu do osób, u których te relacje były mniej wrogie. Dotyczyło to głównie pacjentów z historią zaburzeń nastroju.

Badania wpływu mikrobioty jelitowej na zaburzenia afektywne dwubiegunowe
Ciekawych wniosków dostarczyło badanie w populacji osób chorych na zaburzenia afektywne dwubiegunowe. W trakcie epizodu depresyjnego u tych pacjentów obserwowana była zwiększona liczebność Enterobacteriaceae, podczas gdy u pacjentów w okresie bezobjawowym występowała zwiększona liczebność Clostridiaceae i Roseburia.
Interesujący wydaje się również fakt, że u pacjentów z zaburzeniami afektywnymi dwubiegunowymi w trakcie terapii farmakologicznej stwierdzono wyższą liczebność Klebsiella i Veillonella w porównaniu do pacjentów z nieleczoną chorobą.
Mikrobiota jelitowa a poziom serotoniny
Przykładem tego, jak mikrobiota jelitowa może wpływać na oś mózg-jelita, jest synteza serotoniny, czyli związku zwanego powszechnie „hormonem szczęścia”. Serotonina powstaje w wyniku przemiany tryptofanu. Wiemy, że niedobór serotoniny w ośrodkowym układzie nerwowym jest jednym z czynników wywołujących depresję, smutek, apatię i lęk. Dodatkowo niedobór serotoniny stanowi kluczową rolę w ujawnianiu się obniżonego nastroju. W przewodzie pokarmowym nawet 95% serotoniny jest wytwarzane przez komórki enterochromatofilne (ECC) błony śluzowej, mikroorganizmy wchodzące w skład mikrobioty jelitowej oraz neurony splotów nerwowych w warstwie podśluzowej i mięśniowej jelita. Badania wykazały, że bakterie jelitowe Bifidobacterium infantis wpływają na poziom i metabolizm tryptofanu, zwiększając tym samym jego poziom w organizmie.
Ciekawe zjawisko polegające na stwierdzaniu większej liczebności rodzaju Alistipes zostało zaobserwowane u pacjentów z depresją. Alistipes to bakterie, które zdolne są do konwersji tryptofanu (czyli prekursora serotoniny) w indol. Skutkiem tego jest zmniejszenie stężenia serotoniny w jelicie.
Badania przeprowadzone w ostatnich czterech dekadach wykazały możliwy związek mikrobioty jelitowej z funkcją ośrodkowego układu nerwowego u człowieka, a także potencjalny wpływ na obraz kliniczny chorób psychiatrycznych.
Ciekawych wniosków dostarczyło badanie mające na celu ocenę skuteczności przeszczepu mikrobioty jelitowej w grupie pacjentów z czynnościowymi zaburzeniami przewodu pokarmowego (zespół jelita nadwrażliwego, czynnościowe zaparcia, czynnościowa biegunka). Przeprowadzona analiza sugeruje, że przeszczep mikrobioty jelitowej może łagodzić objawy depresji i lęku niezależnie od wpływu na objawy żołądkowo-jelitowe. Dodatkowo zaobserwowano, że zwiększenie różnorodności mikrobiomu może przyczynić się do poprawy nastroju pacjentów.
Przytoczone fakty jednoznacznie wskazują, że mikrobiota jelitowa to obszar z wielkim potencjałem, którego poznanie może wiązać się z optymalizacją leczenia chorób psychiatrycznych. Prawdopodobnie najbliższe lata i dekady pozwolą nam na lepsze zrozumienie interakcji pomiędzy mózgiem a jelitami, które zamieszkują biliony drobnoustrojów. Być może przyszłość pokaże, że na zaburzenia psychiczne należy patrzeć jako na dysfunkcję zarówno ośrodkowego układu nerwowego, jak i naszego „drugiego mózgu”, czyli jelit.
Piśmiennictwo
- Järbrink-Sehgal E, Andreasson A. The gut microbiota and mental health in adults. Curr Opin Neurobiol. 2020 Jun;62:102-114.
- Peter J, Fournier C, Durdevic M,. A Microbial Signature of Psychological Distress in Irritable Bowel Syndrome. Psychosom Med. 2018 Oct;80(8):698-709
- Skonieczna-Zydecka K, Grochans E, Maciejewska D,. Faecal short chain fatty acids profile is changed in polish depressive women. Nutrients 2018, 10
- Kiecolt-Glaser JK, Wilson SJ, Bailey ML, i wsp.: Marital distress, depression, and a leaky gut: Translocation of bacterial endotoxin as a pathway to inflammation. Psychoneuroendocrinology. 2018 Dec;98:52-60.
- Painold A, Morkl S, Kashofer K, i wsp.: A step ahead: exploring the gut microbiota in inpatients with bipolar disorder during a depressive episode. Bipolar Disord 2019, 21:40-49.
- Rong H, Xie XH, Zhao J i wsp.: Similarly in depression, nuances of gut microbiota: evidences from a shotgun metagenomics sequencing study on major depressive disorder versus bipolar disorder with current major depressive episode patients. J Psychiatr Res 2019, 113:90-99
- Góralczyk-Bińkowska A, Szmajda-Krygier D, Kozłowska E. The Microbiota-Gut-Brain Axis in Psychiatric Disorders. Int J Mol Sci. 2022 Sep 24;23(19):11245.
- Jiang H, Ling Z, Zhang Y i wsp.: Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun 2015; 48: 186–194.
- Song Y, Kononen E, Rautio M: Alistipes onderdonkii sp. nov. and Alistipes shahii sp. nov., of human origin. Int J Syst Evol Microbiol 2006; 56: 1985–1990.
- Cryan J., Dinan T. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012;13:701–712.
- Timothy G. Dinan, Catherine Stanton, John F. Cryan, Psychobiotics: a novel class of psychotropic, „Biological Psychiatry”, 74 (10), 2013, s. 720–726
- Rudzki L, Ostrowska L, Pawlak D, i wsp.: Probiotic Lactobacillus Plantarum 299v decreases kynurenine concentration and improves cognitive functions in patients with major depression: a double-blind, randomized, placebo controlled study. Psychoneuroendocrinology 2019,
- Kurokawa S, Kishimoto T, Mizuno S, i wsp.: The effect of fecal microbiota transplantation on psychiatric symptoms among patients with irritable bowel syndrome, functional diarrhea and functional constipation: an open-label observational study. J Affect Disord 2018