Czekolada to jeden z ulubionych przysmaków tak dzieci, jak i dorosłych. Porcja tego produktu zawiera wiele wartościowych substancji w tym węglowodany, magnez czy potas, które pomagają w szybki sposób uzyskać dodatkową porcję energii w czasie intensywnego wysiłku czy wpływają pozytywnie na nasz układ nerwowy, wspomagając procesy myślowe. Ponadto czekolada może poprawiać nasz nastrój, gdyż związki w niej zawarte podnoszą poziom serotoniny (hormonu szczęścia) w naszym organizmie.
Mimo wszystko czekolada, tak jak większość produktów spożywczych, u niektórych osób może być przyczyną alergii pokarmowej. Związane jest to z patologiczną reakcją układu odpornościowego na kakao – główny składnik czekolady. Ale czy kakao to jedyny winowajca?
Co jeszcze uczula w czekoladzie?
Otóż czekolada to mieszanina różnych alergenów pokarmowych, oprócz samego kakao można znaleźć w niej również:
mleko krowie,
orzechy (włoski, laskowy, orzeszki ziemne),
pszenicę (gluten),
soję.
Wszystkie wymienione źródła alergenowe mogą stanowić poważne zagrożenie dla osób uczulonych i powodować ciężkie objawy alergiczne z reakcją anafilaktyczną włącznie.Mleko jest jedną z najczęstszych przyczyn alergii pokarmowych wśród dzieci. Orzechy bądź ich śladowe ilości można znaleźć w większości czekolad dostępnych na rynku, podobnie jak lecytynę sojową, która używana jest w procesie produkcji czekolady do nadawania jej gładkiej i aksamitnej struktury.
Gluten w czekoladzie?
W przypadku glutenu sprawa nie jest taka prosta. Otóż skład tradycyjnej czekolady nie wskazuje na jego obecność. Jednak przez to, że tradycyjne czekolady wytwarzane są najczęściej w tym samych halach, co czekolady z ciastkami czy innymi dodatkami pochodzenia zbożowego, to może dochodzić do przeniesienia niewielkich ilości tego białka. Dlatego też gorzka czy mleczna czekolada mogą zawierać śladowe ilości glutenu. W przypadku osób uczulonych na gluten czy cierpiących z powodu celiakii zalecamy wybór czekolad z oznaczeniem „produkt bezglutenowy”.
A co z białą czekoladą?
Biała czekolada nie jest do końca czekoladą, ponieważ nie zawiera minimum 35% suchej masy kakaowej. W klasycznej wersji składa się ona z głównie z masła kakaowego (w produktach niższej jakości jest to utwardzony tłuszcz roślinny), mleka oraz cukru. Masło kakaowe to tłuszcz, który nie zawiera białek alergenowych. W związku z tym osoby uczulone na kakao nie powinny doświadczać reakcji alergicznej po spożyciu białej czekolady. Nie należy jednak zapominać o innych uczulających składniach i dodatkach, które mogą znajdować się w tym białym przysmaku.
Objawy alergii na czekoladę
Objawy alergii na składniki czekolady nie odbiegają w znaczący sposób od standardowych objawów alergii pokarmowej. Zalicza się do nich:
pokrzywkę na łokciach, ramionach lub twarzy,
trudności w oddychaniu,
obrzęk języka, warg lub gardła,
świszczący oddech,
ataki astmy,
nudności lub wymioty,
skurcze żołądka.
W przypadku niektórych pacjentów może również dochodzić do anafilaksji.
Diagnostyka uczulenia na czekoladę
W diagnostyce alergii na czekoladę stosuje się zazwyczaj testy IgE z krwi względem składników czekolady i/lub próbę prowokacyjną, która musi zostać wykonana pod okiem wyspecjalizowanego personelu medycznego.
W przypadku badań z krwi sieć laboratoriów ALAB przygotowała dwa pakiety diagnostyczne skomponowane w oparciu o najpowszechniej występujące składniki czekolady. Wszystkie testy uwzględnione w pakietach są wykonywane metodą ImmunoCAP, która uznawana jest za „złoty standard” w diagnostyce alergii in vitro.
Pamiętaj, aby otrzymane wyniki testów skonsultować z lekarzem alergologiem, który zinterpretuje je w oparciu o historię choroby i wyda odpowiednie zalecenia!
Leczenie alergii na czekoladę
Smutna wiadomość dla wszystkich alergików jest taka, że nie ma skutecznego leczenia alergii na czekoladę. Jedynym sposobem uniknięcia objawów alergicznych jest wyłączenie jej z diety. W przypadkach, gdy ryzyko wystąpienia reakcji anafilaktycznej jest duże, lekarz może zalecić zakup automatycznego wstrzykiwacza adrenaliny.
Piśmiennictwo
Lopes JP, Kattan J, Doppelt A, Nowak-Węgrzyn A, Bunyavanich S. Not so sweet: True chocolate and cocoa allergy. J Allergy Clin Immunol Pract. 2019 ; 7(8): 2868–2871
Sadhasivamohan A, Karthikeyan K. Chocolate and Skin: The Impact of an Insatiable Indulgence. Indian Dermatol Online J. 2022 Nov-Dec; 13(6): 806–809.
Vadas P, Perelman B. Presence of undeclared peanut protein in chocolate bars imported from Europe. J Food Prot 2003; 66:1932–4.
Scheibe B, Weiss W, Rueff F, Przybilla B, Gorg A. Detection of trace amounts of hidden allergens: hazelnut and almond proteins in chocolate. J Chromatogr B Biomed Sci Appl 2001; 756:229–37.
Katz DL, Doughty K, Ali A. Cocoa and chocolate in human health and disease. Antioxid Redox Signal 2011; 15:2779–811.
Witamina A to jedna z tych witamin, które pacjenci często suplementują. Jest to de facto grupa związków rozpuszczalnych w tłuszczach, w skład której wchodzi retinol (witamina A1), retinal, kwas retinowy oraz karotenoidy. Głównymi związkami karotenoidów są: β-karoten, α-karoten, β-kryptoksantyna, luteina, likopen.
Najaktywniejszym karotenoidem jest β-karoten będący prowitaminą A. Aby zyskać aktywność biologiczną, musi ulec przekształceniu, dopiero wówczas zyskuje pełną funkcjonalność. Aktywność witaminy A wykazuje również α-karoten i β-kryptoksantyna. Likopen to karotenoid nadający warzywom i owocom barwę czerwoną. Jest silnym antyoksydantem, który – wg badań – zapobiega uszkodzeniom DNA, dzięki czemu może ograniczać rozwój raka prostaty, ma również udział w profilaktyce chorób układu krążenia. Luteina natomiast zapobiega zwyrodnieniu plamki żółtej.
Witamina A spełnia istotną rolę w procesie prawidłowego widzenia, stanowiąc składnik purpury wzrokowej, czyli rodopsyny. Ponadto uczestniczy w prawidłowym różnicowaniu komórek tkanki nabłonkowej i kostnej, bierze udział w produkcji i uwalnianiu hormonów kory nadnerczy i tarczycy. Jest istotnym czynnikiem wspomagającym utrzymanie prawidłowego stanu błon śluzowych i skóry, bierze udział w odczuwaniu smaku. Ponadto witamina A ma potencjał antyoksydacyjny, dzięki czemu wraz z witaminą C i E zapobiega nadmiernemu utlenianiu kwasów tłuszczowych, chroni przed szkodliwym działaniem wolnych rodników, wpływa na prawidłową budowę erytrocytów.
Witamina A bierze udział w tworzeniu plemników, rozwoju łożyska i prawidłowym wzroście płodu.
Istotna rola witaminy A polega również na udziale w utrzymaniu prawidłowej odporności organizmu, pomaga chronić przed infekcjami i je zwalczać.
Biodostępność witaminy A – czynniki wpływające na jej wchłanianie
Witamina A po wchłonięciu z przewodu pokarmowego magazynowana jest przede wszystkim w wątrobie. Nawet do 90% tej substancji zawiera właśnie wątroba, niewielkie ilości są w nerkach, tkance tłuszczowej, osoczu i innych tkankach. Wydalanie witaminy A odbywa się głównie z żółcią.
Na wchłanianie witaminy A mają wpływ różne czynniki. Jednym z nich jest forma witaminy A. Retinol wchłania się w ok. 80%, podczas gdy karotenoidy w ok. 30%.
Wchłanianiu retinolu sprzyja obecność w pokarmie tłuszczów oraz białka. Czynnikiem sprzyjającym jest również obecność kwasów żółciowych oraz prawidłowy poziom hormonów tarczycy.
Czynnikami, które ograniczają jej wchłanianie, są niedostateczna ilość tłuszczu w diecie, alkohol oraz duża ilość błonnika. Przeładowanie organizmu żelazem lub niedobór cynku to również czynniki ograniczające jej wchłanianie.
Witamina A jest wrażliwa na obróbkę kulinarną. Szacuje się, iż straty β-karotenu w czasie gotowania i duszenia warzyw lub owoców mogą wynosić ok. 20%. Przechowywanie warzyw lub owoców w nieprawidłowych warunkach (niewłaściwa temperatura, dostęp światła) dochodzą nawet do 80%. Należy również pamiętać, iż zawartość witaminy A w warzywach i owocach uzależniona jest również od pory roku.
Również obróbka kulinarna mięsa czy ryb powoduje 20% straty tej witaminy. Duszenie, pieczenie, gotowanie, smażenie to procesy zmniejszające zawartość witaminy A w przyjmowanych pokarmach.
Zapotrzebowanie i źródła witaminy A
Witamina A jest obecna zarówno w pokarmach roślinnych, jak i produktach pochodzenia zwierzęcego. Rośliny są źródłem karotenoidów, zwłaszcza β-karotenu, czyli prowitaminy A, a mięso retinolu i jego pochodnych.
Głównym źródłem witaminy A dla Polaków są mleko i jego przetwory, jaja, wątróbka i podroby, ryby.
Objawy niedoboru i nadmiaru witaminy A w organizmie
Głównym źródłem witaminy A w diecie Polaków są produkty zwierzęce – 60%, z kolei 40% pochodzi z produktów roślinnych. Niedobory tej witaminy występują stosunkowo rzadko, objawy pojawiają się dopiero po wyczerpaniu jej zapasów w wątrobie.
Wczesnym symptomem niedoboru jest suchość skóry i nadmierne rogowacenie naskórka. Następne objawy to zaburzenia występujące w nabłonku dróg oddechowych, przewodu pokarmowego, dróg moczowych i pochwy. Zmniejsza się wydzielanie śluzu, jego działanie ochronne jest niewystarczające, co może skutkować zmniejszoną odpornością na zakażenia bakteryjne. Charakterystycznym objawem niedoboru witaminy A jest kurza ślepota, czyli tzw. niedowidzenie zmierzchowe.
Pozostałe objawy to łamliwość włosów i paznokci, zaburzenia łaknienia, problemy z zajściem w ciążę, zaburzenia wzrostu kości i zębów, osteoporoza.
Nadmierna podaż witaminy A występuje głównie wskutek niekontrolowanej suplementacji. Może przyczyniać się do: drażliwości i zaburzeń koncentracji, bólów głowy, nudności, wymiotów, światłowstrętu.
Nadmierna podaż witaminy A u kobiet w ciąży może prowadzić do zaburzeń rozwoju płodu.
Piśmiennictwo
Ostrowska L., Diagnostyka laboratoryjna w dietetyce, PZWL 20182.
Włodarek D., Lange E., Kozłowska L., Głąbska D, Dietoterapia, PZWL 2014
Pierwotne zapalenie dróg żółciowych – PBC (primary biliary cholangitis) dawniej pierwotna marskość żółciowa PBC (primary biliary cirrhosis) – nazwa choroby uległa zmianie, ponieważ u większości chorych z rozpoznaniem tej choroby nie stwierdza się cech zaawansowanego zwłóknienia wątroby, które nazywamy właśnie marskością. Jest to postępujący zespół cholestatyczny o nieustalonej ostatecznie etiologii, występujący głównie u kobiet pomiędzy 40. a 60. rokiem życia (najczęściej ok.50 r.ż.). Schorzenie ma charakter przewlekły, zapalny o podłożu autoimmunizacyjnym.
Zapadalność na pierwotne zapalenie dróg żółciowych(PBC) u kobiet i mężczyzn występuje w proporcji odpowiednio ok. 10:1. Choroba nie występuje u dzieci. Obserwuje się pewną regionalność zachorowań. Pierwotne zapalenie dróg żółciowych częściej pojawia się w miastach oraz w państwach znajdujących się na półkuli północnej. Istnieją doniesienia na temat wpływu czynników genetycznych na rozwój schorzenia – częściej choroba występuje u córek matek z pierwotnym zapaleniem dróg żółciowych (PBC), dawniej zwanym pierwotną marskością żółciową.
Objawy pierwotnego zapalenia dróg żółciowych
Objawy kliniczne są mało charakterystyczne, dlatego rozpoznanie zazwyczaj ustalane jest przypadkowo. Pacjenci skarżą się najczęściej na uczucie przewlekłego zmęczenia o stałym natężeniu, bez względu na stan psychofizyczny. U chorych występuje również uporczywy świąd skóry, przeczosy na skórze, przebarwienia skóry, u niektórych kępki żółte – tzw. żółtaki (zmiany skórne w okolicach oczodołów), rzadko występuje żółtaczka. Pojawia się dyskomfort pod prawym łukiem żebrowym, u około 25% pacjentów hepato- i splenomegalia, często bezsenność, zaburzenia poznawcze oraz depresja.
Do objawów współistniejących z innymi chorobami autoimmunizacyjnymi zalicza się: suchość błon śluzowych, bóle stawowe, niedoczynność tarczycy. Może wystąpić również osteoporoza będąca wynikiem upośledzonego wchłaniania witaminy D3 na tle przewlekłej cholestazy, niedobory pozostałych witamin rozpuszczalnych w tłuszczach (wit. A, E i K) oraz zaburzenia lipidowe.
Istotą choroby jest postępujący proces uszkadzania małych i średnich przewodów żółciowych wewnątrzwątrobowych prowadzący do rozwoju cholestazy, a w efekcie do niewydolności wątroby. Biologia i funkcjonowanie hepatocytów w początkowej fazie choroby pozostają niezaburzone.
Przyczyny pierwotnego zapalenia dróg żółciowych
Jedną z koncepcji powstawania pierwotnego zapalenia dróg żółciowych (PBC) jest uszkodzenie prawidłowo funkcjonujących cholangiocytów przez konkretne czynniki autoimmunizacyjne, infekcje i uszkadzające działanie toksyn wydzielanych wraz z żółcią. Oddziaływanie tych czynników prowadzi do uszkodzenia cholangiocytów, a w rezultacie do uszkodzenia przewodów żółciowych.
Dotychczas nie odkryto konkretnego czynnika inicjującego aktywację układu immunologicznego w kierunku niszczenia cholangiocytów dróg żółciowych. Za podłożem autoimmunologicznym choroby może przemawiać zwiększona częstość występowania PBC u pacjentów dotkniętych innymi chorobami z autoagresji.
Badacze zwracają również uwagę na wpływ zaburzeń ekspresji w układzie ludzkich antygenów leukocytarnych (HLA – human leukocyte antigens) na rozwój pierwotnego zapalenia dróg żółciowych (PBC). Patologie HLA mogą generować podatność tkanek na atak cytotoksycznych limfocytów T, a nadekspresja antygenów HLA-DR w cholangiocytach dróg żółciowych powoduje, że układ odpornościowy rozpoznaje te komórki jako prezentujące antygen. Przez taką zmianę fenotypu komórkowego cholangiocyty stają się celem ataku komórek cytotoksycznych. Uszkodzenie nabłonka dróg żółciowych może być również wynikiem występującej w nim zmiany ekspresji antygenów. Konsekwencją tego jest zwiększona odpowiedź immunologiczna z uwolnieniem cytokin, między innymi: interleukiny 2, interleukiny 12,interferonu gamma, TNF α.
Niektórzy badacze zwracają uwagę na wpływ dysbiozy jelitowej oraz translokacji mikrobioty jelitowej do wątroby jako czynnika promującego rozwój pierwotnego zapalenia dróg żółciowych (PBC). Mechanizmy pobudzenia autoagresji przez translokację mikroflory jelitowej pozostają nieznane. Dodatkowo podkreśla się czynnik infekcyjny rozwoju PBC. Infekcja układu moczowego wywołana przez bakterię Escherichia coli może zwiększać ryzyko rozwoju PBC.
Diagnostyka pierwotnego zapalenia dróg żółciowych
Kryteria diagnostyczne pierwotnego zapalenia dróg żółciowych (PBC) opierają się na rozpoznaniu zespołu cholestatycznego utrzymującego się przez 6 miesięcy z podwyższoną aktywnością ALP (fosfatazy alkalicznej) i GGTP (gamma – glutamylotranspeptydazy), obecnością autoprzeciwciał mitochondrialnych oraz charakterystycznych zmian zapalnych przewodów żółciowych, którego skutkiem jest utrudniony odpływ żółci oraz postępująca niewydolność wątroby. Podwyższone stężenie bilirubiny w momencie rozpoznania, podobnie jak postępujący wzrost stężenia tego parametru, wskazuje na zaawansowane stadium choroby i złe rokowanie. U ponad połowy pacjentów stwierdza się zaburzenia lipidowe i niedobory witamin rozpuszczalnych w tłuszczach. Zwiększone jest również stężenie immunoglobulin IgM i IgG.
Diagnostyka laboratoryjna
aktywność fosfatazy zasadowej – ALP(alkaline phosphatase) w surowicy co najmniej 1,5 x wyższa od górnej granicy wartości referencyjnych (wzrost aktywności ALP koreluje z nasileniem duktopenii – uszkodzeniem/zanikiem drobnych dróg żółciowych i progresją choroby),
5 krotne przekroczenie wartości referencyjnych ALT wymaga różnicowania z zespołem nakładania PBC i AIH,
Lipidogram– u 50% pacjentów występuje hipercholesterolemia, zwiększone stężenie głównie cholesterolu LDL oraz Lpx (lipoproteiny X – czuły wskaźnik cholestazy),
Wyróżnia się obecnie 9 typów AMA (M1-M9) reagujących z różnymi białkami mitochondrialnymi, głównie enzymami. W surowicy pacjentów z pierwotnym zapaleniem dróg żółciowych (PBC) dotychczas wykryto 4 typy AMA: przeciwciała przeciw antygenom M2, M4, M8, oraz M9.
Przeciwciała AMA typu 2 (AMA-M2) reagują z podjednostkami kompleksu dehydrogenazy pirogronianowej (PDH-E2), dehydrogenazy α- ketokwasów (BCOADH-E2) i dehydrogenazy α-ketoglutaranu (OGDH). U większości pacjentów (80-90%) wykrywa się p/c AMA-M2-PDH-E2, dlatego też za główny autoantygen w PBC uznaje się składową E2 dehydrogenazy pirogronianowej. Część pacjentów (około 60%) reaguje również z BCOADH-E2.
Przeciwciała AMA typ 4 (AMA-M4) reagują z oksydazą tiolową, AMA typ 9 (AMA-M9) z fosforylazą glukanową. Zwykle występują z AMA-M2.
Przeciwciała AMA-M2 są swoistym i czułym markerem diagnostycznym PBC, wykrywa się je u ok. 94% pacjentów, stanowią też narzędzie prognozujące późniejszy rozwój schorzenia u pacjentów bez znaczących zaburzeń funkcji wątroby sugerujących chorobę cholestatyczną.
Przeciwciała AMA-M2 mogą występować również w innych chorobach nakładających się na pierwotne zapalenie dróg żółciowych (PBC) np. w AIH, ale również w sklerodermie czy zespole Sjögrena. Zasadniczo w innych chorobach, poza PBC, przeciwciała AMA występują w niskich mianach. Istotne klinicznie jest miano AMA ≥1:40.
Przeciwciała przeciwjądrowe (ANA)
Drugą grupą przeciwciał wykorzystywaną w diagnostyce są przeciwciała przeciwjądrowe – ANA(antinuclear antibodies) – anty-Sp100 i anty-gp210, które charakteryzują się niską czułością oraz występują tylko u około 30% pacjentów z pierwotnym zapaleniem dróg żółciowych (PBC), niemniej mają wysoką swoistość w odniesieniu do tej jednostki chorobowej – ok. 95%. W przebiegu PBC najczęściej obserwuje się typ świecenia „nuclear dots”, za który odpowiadają między innymi przeciwciała Sp100, oraz typ świecenia „błony jądrowej”, związany z przeciwciałami przeciwko gp210.
Kryteria rozpoznania pierwotnego zapalenia dróg żółciowych
American Association for the Study of Liver Diseases (AASLD) w 2018 roku opublikowało zaktualizowane wytyczne, w których podstawę rozpoznania PBC stanowi stwierdzenie u pacjenta dwóch z trzech niżej wymienionych cech klinicznych:
podwyższona aktywność ALP w surowicy (>1,5 × górna granica wartości referencyjnych),
obecność AMA w mianie ≥1:40,
przewlekłe nieropne uszkodzenie dróg żółciowych w bioptacie wątroby.
W przypadku niewystępowania przeciwciał AMA pozytywny wynik oznaczenia przeciwciał anty-Sp100 i anty-gp210 potwierdza rozpoznanie pierwotnego zapalenia dróg żółciowych (PBC).
Zgodnie z rekomendacjami EASL (European Association for the Sudy of the Liver) u pacjentów z podwyższonym poziomem ALP i obecnością przeciwciał AMA w wysokim mianie (≥1:40) diagnozę PBC można postawić bez wykonywania biopsji wątroby.
Leczenie pierwotnego zapalenia dróg żółciowych
W leczeniu pierwotnego zapalenia dróg żółciowych stosuje się początkowo postępowanie niefarmakologiczne, które obejmuje umiarkowany wysiłek fizyczny i regularne ćwiczenia (mogą zmniejszyć uczucie przewlekłego zmęczenia oraz zmniejszają ryzyko osteopenii) oraz leczenie objawowe np. świądu skóry przez stosowanie emolientów czy odpowiednich kąpieli. Natomiast leczenie farmakologiczne polega głównie na stosowaniu kwasu ursodeoksycholowego i kwasu obeticholowego. Eksperci EASL w maju 2017 r. dopuścili kwas obeticholowy (OCA – obeticholic acid) do stosowania w połączeniu z UDCA u pacjentów z pierwotnym zapaleniem dróg żółciowych (PBC), u których stwierdza się niewystarczającą odpowiedź na UDCA, lub jako monoterapię u osób, które nie tolerują UDCA.
Po roku od wdrożenia terapii dokonuje się oceny jej skuteczności, wykonując badania biomarkerów wątrobowych w surowicy. Najczęściej są wykorzystywane tzw. kryteria barcelońskie definiujące pozytywną odpowiedź na leczenie UDCA poprzez spadek aktywności ALP o więcej niż 40% wartości wyjściowej lub jej całkowitą normalizację. Badania dowodzą, że największą korzyść z leczenia UDCA odnoszą pacjenci, którzy w chwili wdrożenia terapii znajdowali się we wcześniejszych stadiach choroby.
Pacjenci z pierwotnym zapaleniem dróg żółciowych (PBC) są narażeni na zwiększone ryzyko rozwoju raka wątrobowokomórkowego (HCC –hepatocellular carcinoma), chociaż w mniejszym stopniu niż osoby z innymi przewlekłymi chorobami wątroby. Do czynników ryzyka rozwoju HCC w pierwotnym zapaleniu dróg żółciowych są zaliczane: zaawansowany wiek i stadium choroby w momencie rozpoznania, płeć męska oraz brak lub niewielka odpowiedź biochemiczna na terapię UDCA.
Piśmiennictwo
EASL Clinical Practice Guidelines: management of cholestatic liver diseases, Hepatol 2009, 51 (2).
K.Buczkowski, L.Wylężałek, M.Dyaczyński, M.Waluga: Pierwotne zapalenie dróg żółciowych, Medycyna po Dyplomie 2021,01.
Y.Shoenfeld, P.L Meroni: The general practice guide to autoimmune disease, Pabst science publishers Lengerich 2012.
GM Hirschfield , JK Dyson , GJM Alexander, et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut 2018
HD Ma, ZB Zhao , WT Ma , et al. Gut microbiota translocation promotes autoimmune cholangitis. J Autoimmun 2018.
F Nevens , P Andreone , G Mazzella , et al. POISE Study Group. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016
P Invernizzi , A Lleo , M Podda . Interpreting serological tests in diagnosing autoimmune liver diseases. Semin Liver Dis 2007.
Zespół antyfosfolipidowy (antiphospholipid syndrome – APS) to niezapalna, układowa choroba tkanki łącznej o podłożu autoimmunizacyjnym. Jest najczęściej występującą, nabytą trombofilią (nadkrzepliwością) i powoduje: zakrzepicę żylną i tętniczą o różnej lokalizacji (która występuje u 59% chorych w naczyniach żylnych, u 28% w naczyniach tętniczych, u 13% chorych zarówno w naczyniach żylnych, jak i tętniczych), małopłytkowość wtórną, przyczynia się do niepowodzeń położniczych, porodów przedwczesnych u pacjentek z preeklampsją (w stanach przedrzucawkowych).
W zespole antyfosfolipidowym produkowane są autoprzeciwciała (antiphospholipid autoantibodies – APL) przeciw ujemnie naładowanym fosfolipidom błon komórkowych, co prowadzi do zaburzeń hemostazy o charakterze prozakrzepowym. Są to głównie przeciwciała przeciw kardiolipinowe (aCL), przeciw β-2 glikoproteinie – I (β-2GP I ) oraz LA – antykoagulant tocznia. Przeciwciała te charakteryzują się wystarczająco wysoką czułością i swoistością, dlatego mają charakter diagnostyczny.
W jaki sposób przeciwciała antyfosfolipidowe zwiększają ryzyko zakrzepicy i niepowodzeń położniczych?
Mechanizmy indukujące zmiany zakrzepowe w naczyniach obejmują aktywację płytek krwi i komórek śródbłonka przez wiązanie się przeciwciał antyfosfolipidowych z fosfolipidami błony komórkowej oraz białkami osocza, tworzącymi kompleksy z fosfolipidami głównie β2-glikoproteiną I oraz białkami biorącymi udział w procesie krzepnięcia protrombiną, aneksyną 5 i białkiem C, białkiem S, kininogenami, czynnikami układu krzepnięcia (VII, XI, XII), składowymi układu dopełniacza (C4, czynnik H).
Proces patologiczny zaczyna się od aktywacji płytek krwi, komórek śródbłonka lub komórek trofoblastu. Dochodzi wtedy do przemieszczenia ujemnie naładowanego fosfolipidu fosfatydyloseryny z wnętrza komórki na jej powierzchnię, która w warunkach fizjologicznych jest elektrycznie obojętna. Z fosfatydyloseryną wiąże się krążąca w surowicy β2-glikoproteina I. Z kompleksem β2-GPI-fosfatydyloseryna wiążą się następnie przeciwciała antyfosfolipidowe i następuje aktywacja dopełniacza drogą klasyczną, co z kolei indukuje ekspresję molekuł adhezyjnych, czynnika tkankowego, oraz aktywuje monocyty, granulocyty i płytki krwi. W konsekwencji uwalniane są mediatory zapalenia ( TNFα, VEGR-1) i zwiększa się ryzyko zakrzepicy. W tym czasie mogą również wystąpić zmiany zaburzające procesy prawidłowego rozwoju łożyska.
Mechanizmy działania przeciwciał antyfosfolipidowych
1. Hamowanie funkcji naturalnych inhibitorów krzepnięcia:
hamowanie aktywności przeciwzakrzepowej β2- GPI
hamowanie aktywacji białka C
hamowanie funkcji aktywowanego białka C
hamowanie aktywności antytrombiny
przemieszczenie aneksyny V
2. Modyfikacja czynności komórek:
a) Śródbłonka
indukcja ekspresji czynnika tkankowego
indukcja ekspresji cząsteczek adhezyjnych
obniżenie produkcji prostacykliny
pobudzenie produkcji tromboksanu płytkowego
b) Monocytów:
indukcja ekspresji czynnika tkankowego
c) Płytek krwi
wzrost aktywacji lub agregacji
Objawy zespołu antyfosfolipidowego (APS)
Do najczęstszych objawów zespołu antyfosfolipidowego należą objawy neurologiczne. Obserwowane są zarówno objawy ze strony ośrodkowego, jak i obwodowego układu nerwowego. Są to: udar mózgu oraz przemijające niedokrwienie (transient ischaemic attack – TIA), padaczka, pląsawica, bóle głowy, poprzeczne zapalenie rdzenia oraz obwodowa polineuropatia.
W przebiegu zespołu antyfosfolipidowego (APS) mogą się pojawiać także zaburzenia psychiczne. Ich etiologia nie jest jasna. Przyjmuje się, że mogą one być wtórne do niedokrwienia tkanki mózgowej w procesie zakrzepowym towarzyszącym tworzeniu APL i/lub są następstwem bezpośredniej interakcji obecnych w krążeniu przeciwciał antyfosfolipidowych z tkanką nerwową. Najczęściej obserwuje się zaburzenia funkcji poznawczych charakteryzujące się pogorszeniem uwagi, koncentracji, zaburzenia depresyjne, oraz zespoły demencyjne.
ASP jest również poważnym problemem perinatologicznym, który wiąże się z nawracającymi stratami ciąż, wewnątrzmacicznym opóźnieniem wzrostu płodu, stanem przedrzucawkowym i ablacją łożyska.
Kryteria rozpoznania zespołu antyfosfolipidowego (APS)
Kryteria laboratoryjne:
Antykoagulant tocznia LA obecny w osoczu i wykryty >= 2 krotnie w odstępach 12 tygodni metodami ustalonymi przez MTZH.
Obecność przeciwciał antykardiolipinowych w klasie IgG (GLP) i/lub IgM (MPL)w surowicy lub osoczu w średnim, lub dużym stężeniu (>40 GPL lub MPL albo > 99 percentyla) stwierdzona minimum dwukrotnie w odstępie 12 tygodni, pomiar z użyciem standaryzowanego testu ELISA.
Międzynarodowy zespół ekspertów zaleca wprowadzenie podziału chorych na zespół antyfosfolipidowy (APS) zależnego od kryteriów laboratoryjnych:
Grupa I: obecne> niż jedno kryterium laboratoryjne (dowolna kombinacja)
Grupa IIa: obecny tylko LA
Grupa IIb: obecne tylko aCL
Grupa IIc: obecne tylko aβ2-GPI
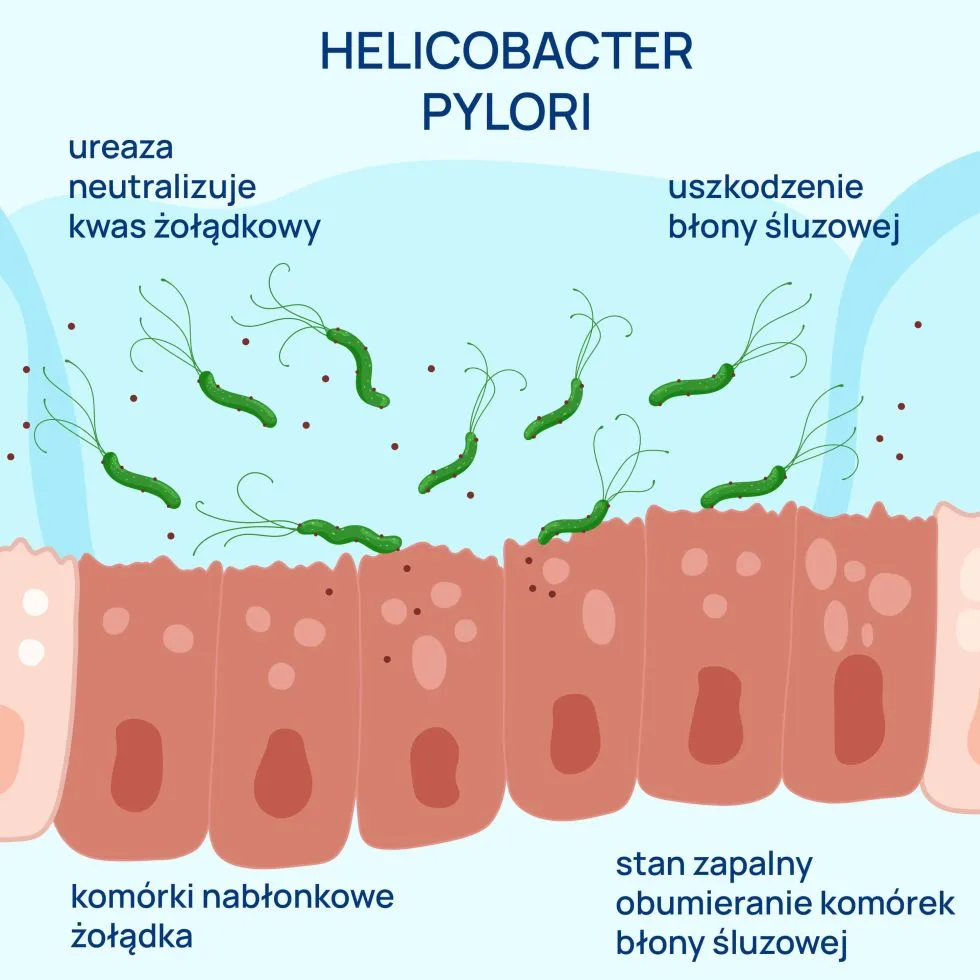
Dwukrotne oznaczenie aPL jest związane z obecnością tych przeciwciał również w przebiegu infekcji bakteryjnych (kiła, borelioza, trąd, gruźlica, zakażenia gronkowcem, paciorkowcem, Helicobacter pylori, Klibsiella, Mycoplasma pneumonie), wirusowych (ospa, różyczka, zakażeniach HIV, CMV, HBV i HCV), grzybiczych lub pasożytniczych (malaria, toksoplazmoza). Przeciwciała te najczęściej nie dają objawów klinicznych i zanikają po kilku tygodniach.
Przeciwciała antyfosfolipidowe można również zaobserwować w chorobach neurologicznych (stwardnienie rozsiane, padaczka, pląsawica) oraz w schyłkowej niewydolności nerek. Mogą być również indukowane przez leki np. chloropromazynę, prokainamid, hydralazynę, chininę, chinidynę, streptomycynę oraz doustne leki antykoncepcyjne.
Czynniki ryzyka rozwoju zespołu antyfosfolipidowego (APS)
palenie papierosów
okres okołooperacyjny i przedłużające się unieruchomienie
obecność aPL z towarzyszącym polimorfizmem czynnika V Leiden
wrodzone niedobory czynników krzepnięcia
hiperhomocysteinemia
Przeciwciała antyfosfolipidowe mogą utrzymywać się w wysokim mianie latami, nie dając żadnych objawów, do ujawnienia się ich działania prozakrzepowego potrzebny jest dodatkowy czynnik sprawczy np. uszkodzenie naczyń, aktywacja śródbłonka lub występowanie czynników ryzyka wyżej wymienionych.
Przeciwciała antyfosfolipidowe przydatne w diagnostyce zespołu antyfosfolipidowego (APS), a nie należące do kryteriów rozpoznania (klasyfikacji)
U niektórych chorych nie stwierdza się też obecności przeciwciał aCL i aβ2-GPI w klasach IgG i IgM, ale mogą być one obecne w klasie IgA.
W przypadkach, gdy charakterystycznym objawom zespołu antyfosfolipidowego (APS) nie towarzyszy obecność przeciwciał antyfosfolipidowych, należy wykluczyć przede wszystkim laboratoryjne przyczyny wyników ujemnych, ocenić inne, nieujęte w kryteriach aPL (jeśli wyniki są dodatnie, rozpoznać zespół antyfosfolipidowy), gdy wyniki są ujemne, można rozpoznać zespół antyfosfolipidowy (APS) seronegatywny SN-APS, albo rozważyć inne możliwe tło zakrzepicy.
Zespół antyfosfolipidowy może być samoistny – pierwotny (primary APS – PAPS) lub towarzyszyć innym chorobom – wtórny zespół antyfosfolipidowy (secondary APS- SAPS). Najczęściej są to układowe choroby tkanki łącznej, głównie toczeń rumieniowaty układowy (SLE – systemic lupus erythematosus), ale również nowotwory i zakażenia. Opisano również wyraźną predyspozycję rodzinną do występowania choroby. Częstość występowania zespołu jest istotnie wyższa wśród chorych z: SLE- ok.30%, zakrzepicą żylną – 30%, udarem mózgu <50 roku życia – 25%, nawracającymi niepowodzeniami położniczymi – 10%. Kobiety chorują częściej niż mężczyźni (3,5: 1).
Przeciwciała antyfosfolipidowe mogą występować w niskim mianie u osób zdrowych (uczestniczą w eliminacji oksydowanych lipidów). Występują u 1-5% populacji ogólnej, a częstość ich występowania rośnie z wiekiem. Przeciwciała antykardiolipinowe wykazuje się w 2-5% u osób w średnim wieku i nawet do 50% u osób powyżej 70 lat, natomiast LA stwierdza się tylko u 1% osób zdrowych.
Piśmiennictwo
M. Puszczewicz red.: Wielka Interna Reumatologia, Medical Tribune, Warszawa 2020.
K. Fiszer, M. Brzosko: Diagnostyka laboratoryjna chorób reumatycznych, Wyd. PUM. Szczecin 2015.
M. Majdan , A. Majdan: Układowe choroby tkanki łącznej przebiegające z zaburzeniami psychicznymi. Psychiatria po Dyplomie 2014; 11: 20-24.
A. Marszałek: Zespół antyfosfolipidowy – współczesne kryteria rozpoznawania, Diagn Lab 2015; 51: 63-66.
B. Grygiel-Górniak, N. Limphaibool, M. Puszczewicz : Cytokine secretion and the risk of depression development in patients with connective tissue diseases. Psychiatr Clin Neurosci 2019; 73: 302-316.
A. Lorenc, A. Seremak-Mrozikiewicz, K. Drews i wsp.: A case of antiphospholipid syndrome in course of pregnancy and puerperium with lupus-like disease suspicion. Ginekol Pol 2011; 82: 297-303.
Metformina jest lekiem przeciwcukrzycowym. Należy do grupy biguanidów, których działanie znane jest już od lat dwudziestych XX wieku. Spośród całej tej grupy jedynie metformina zachowała zastosowanie lecznicze.
Stosowanie doustnych leków obniżających poziom cukru zalecane jest gdy:
leczenie dietetyczne i redukcja masy ciała nie przynoszą oczekiwanych rezultatów,
nie ma wskazań do stosowania insuliny zamiast doustnych leków przeciwcukrzycowych.
W przypadku nie tylko metforminy, ale wszystkich doustnych leków obniżających poziom cukru trzeba pamiętać, że stosuje się je, dopiero gdy zmiana stylu życia tj. zmiana nawyków żywieniowych oraz wprowadzenie regularnego wysiłku fizycznego nie przynosi efektów.
Polskie Towarzystwo Diabetologiczne zaleca rozpoczęcie leczenia metforminą, dopiero gdy mimo stosowania diety i wysiłku fizycznego nie osiągnięto wyrównania poziomu cukru przez 4 tygodnie. Metformina staje się lekiem pierwszego wyboru dla powyżej opisanych pacjentów zwłaszcza z otyłością (szczególnie w przypadku otyłości brzusznej) lub z nadwagą oraz osób z zaburzeniami profilu lipidowego.
Metformina – działanie
Do tej pory ustalono, że metformina działa na kilku płaszczyznach wywołując efekt przeciwcukrzycowy, mianowicie:
hamuje wchłanianie glukozy w środkowej części jelita cienkiego,
zmniejsza produkcję glukozy w wątrobie, co wynika z hamowania wątrobowego procesu glukoneogenezy. Dokładny mechanizm tego procesu nie został jeszcze poznany, ale przypuszcza się, że miejscem jej działania są mitochondria hepatocytów, w których metformina hamuje aktywność kompleksu I łańcucha oddechowego,
zwiększa wrażliwość tkanek obwodowych na działanie insuliny – o tym dokładnie w następnym akapicie,
zwiększa syntezę oraz wpływa na aktywność transporterów glukozy – GLUT 1 i GLUT 4 w komórkach mięśni szkieletowych. Niedostateczna ilość tych transporterów powoduje zmniejszony wychwyt glukozy zależny od insuliny, który prowadzi do zahamowania transportu glukozy,
obniża stężenie wolnych kwasów tłuszczowych (WKT) – zwłaszcza u pacjentów chorych na cukrzycę typu 2 zaobserwowano podwyższone stężenie WKT. Liczne badania donoszą również, że wzrost stężenia WKT jest czynnikiem przyspieszającym rozwój choroby. Prawdopodobnie dzieje się tak z powodu hamowania pobudzanego insuliną wychwytu glukozy w mięśniach i upośledzenie wydzielania insuliny przez komórki β-trzustki. U pacjentów z cukrzycą typu 2 metformina zmniejsza utlenianie WKT według różnych źródeł średnio o 25%,
nasila glikolizę beztlenową i wytwarzanie mleczanów poprzez zmniejszanie wewnątrzkomórkowej produkcji ATP a tym samym zwiększa zużycie glukozy,
inne badania donoszą, że aktywacja AMPK przez metforminę hamuje ekspresję genów kodujących enzymy lipogenezy, czyli mówiąc wprost procesu syntezy tkanki tłuszczowej.
Metformina a witamina B12
Mimo że jest to lek o bardzo dobrych efektach terapeutycznych, wielu pacjentów zwłaszcza w początkowym etapie leczenia skarży się na pojawiające się działania niepożądane. Pocieszające wydaje się, że wraz z czasem trwania leczenia efekty te słabną, aż w końcu całkowicie zanikają. Szacuje się, że tylko u ok. 5% pacjentów rozpoczynających kurację metforminą, działania niepożądane są na tyle uciążliwe, że wymagają przerwania kuracji.
Jednym z najczęściej przytaczanych działań niepożądanych jest wpływ leczenia metforminą na niedobór witaminy B12.
Uważa się że u 5,8% do 33% osób stosujących metforminę występuje obniżony poziom witaminy B12. Prawdopodobnie dzieje się tak z powodu:
pobudzenia przerostu flory bakteryjnej jelita cienkiego, prowadzącego do zaburzenia trawienia i wchłaniania,
hamowania lub całkowitego blokowania absorpcji witaminy B12 poprzez zmniejszenie ilości IF (intrinsic factor, czyli czynnika, który w żołądku tworzy kompleks z wit. B12 odporny na trawienie i umożliwia jej wchłanianie w jelitach),
hamowania wchłaniania kompleksu witaminy B12 z czynnikiem IF w jelicie.
W konsekwencji tego mogą wystąpić niedobory witaminy B12 objawiające się:
zaburzenia w układzie krwionośnym (związane głównie z niedostateczną produkcją krwinek czerwonych): niedokrwistość złośliwa, niedokrwistość megaloblastyczna,
objawy towarzyszące rozwijającej się anemii: osłabienie, zmęczenie, zaburzenia pamięci i koncentracji, bóle i zawroty głowy,
zaburzenia w układzie nerwowym: uczucie mrowienia w rękach czy stopach, uczucie prądu przechodzącego wzdłuż kręgosłupa, osłabienie wzroku,
zmiany zwyrodnieniowe błony śluzowej żołądka oraz zaburzenia wchłaniania,
bolesne owrzodzenia w kącikachust,
hiperhomocysteinemia, co z kolei predysponuje do rozwoju chorób układu krążenia.
W związku z tym zasadne wydaje się być rutynowe, okresowe sprawdzanie poziomu witaminy B12 u pacjentów przewlekle stosujących metforminę, pomoże to uniknąć wystąpienia jej niedoboru poprzez wprowadzenie właściwej suplementacji.
Piśmiennictwo
Zalecenia kliniczne dotyczące postępowania u chorych na cukrzycę. 2020, Stanowisko Polskiego Towarzystwa Diabetologicznego.
Grzybowska M., Bober J., Olszewska M.: Metformina – mechanizmy działania i zastosowanie w terapii cukrzycy typu 2. Postepy Hig Med Dosw (online), 2011; 65: 277-285.
Andres E., Noel E., Goichot B. Metformin-associated vitamin B12 deficiency. Arch. Int. Med. 2002; 162: 2251–2252.
Kos E., Liszek M.J., Emanuele M.A., Durazo-Arvizu R., Camacho P. Effect of metformin therapy on vitamin D and vitamin B12 levels in patients with type 2 diabetes mellitus. Endocr Pract. 2012; 18: 179–184.
de Jager J., Kooy A., Lehert P. i wsp. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ 2010; 340: c2181.
Gumprecht J., Długaszek M., Niemczyk A., Pyryt M., Olszańska E., Gubała M., Tyrała K., Kwiendacz H., Nabrdalik K.: Czy należy obawiać się niedoborów witaminy B12 w trakcie leczenia metforminą? Diabetologia Praktyczna 2016, tom 2, nr 6.
Rozporządzenie Ministra Zdrowia z dnia 16 września 2010 r. w sprawie substancji wzbogacających dodawanych do żywności. Dz. U. 2010 r. Nr 174, poz. 1184.
Kräutler B.: The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Subcell Biochem. 2012;56:323-46.
Banerjee R., Ragsdale S.W.: The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209-47.
Giedyk M., Goliszewska K., Gryko D.: Vitamin B12 catalysed reactions. Chem Soc Rev. 2015 Jun 7;44(11):3391-404.
Paul C., Brady D. M.: Bioavailability and Utilization of Particular Forms of B12 Supplements With Potential to Mitigate B12-related Genetic Polymorphisms. Integr Med (Encinitas). 2017 Feb;16(1):42-49.
Shane B.: Folate and vitamin B12 metabolism: overview and interaction with riboflavin, vitamin B6, and polymorphisms. Food and nutrition bulletin, 2008, 29.2_suppl1: S5-S16.
Andres E., Noel E., Goichot B. Metformin-associated vitamin B12 deficiency. Arch. Int. Med. 2002; 162: 2251–2252.
Kos E., Liszek M.J., Emanuele M.A., Durazo-Arvizu R., Camacho P. Effect of metformin therapy on vitamin D and vitamin B12 levels in patients with type 2 diabetes mellitus. Endocr Pract. 2012; 18: 179–184.
de Jager J., Kooy A., Lehert P. i wsp. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ 2010; 340: c2181.
Gumprecht J., Długaszek M., Niemczyk A., Pyryt M., Olszańska E., Gubała M., Tyrała K., Kwiendacz H., Nabrdalik K.: Czy należy obawiać się niedoborów witaminy B12 w trakcie leczenia metforminą? Diabetologia Praktyczna 2016, tom 2, nr 6.
Rozporządzenie Ministra Zdrowia z dnia 16 września 2010 r. w sprawie substancji wzbogacających dodawanych do żywności. Dz. U. 2010 r. Nr 174, poz. 1184.
Kräutler B.: The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Subcell Biochem. 2012;56:323-46.
Banerjee R., Ragsdale S.W.: The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209-47.
Giedyk M., Goliszewska K., Gryko D.: Vitamin B12 catalysed reactions. Chem Soc Rev. 2015 Jun 7;44(11):3391-404.
Paul C., Brady D. M.: Bioavailability and Utilization of Particular Forms of B12 Supplements With Potential to Mitigate B12-related Genetic Polymorphisms. Integr Med (Encinitas). 2017 Feb;16(1):42-49.
Shane B.: Folate and vitamin B12 metabolism: overview and interaction with riboflavin, vitamin B6, and polymorphisms. Food and nutrition bulletin, 2008, 29.2_suppl1: S5-S16.
Dieta eliminacyjna to czasowe lub stałe usunięcie z żywienia źle tolerowanych produktów przy jednoczesnym wprowadzeniu składników o podobnych właściwościach odżywczych.
Jest to trudny i wymagający proces dlatego decyzja o diecie eliminacyjnej powinna zostać podjęta w oparciu o obserwacje kliniczne, wyniki badań (próba prowokacji i eliminacji, IgE specyficzne dla alergenów pokarmowych, testy skórne) i wywiad.
Zawsze należy zestawić wyniki z objawami. W tym celu niezbędne jest prowadzenie dzienniczków dietetycznych, w których pacjenci zapisują spożywane produkty i ewentualne pojawienie się zaostrzenia zmian skórnych.
Jeśli widać związek pomiędzy spożywanymi produktami a zaostrzeniem objawów można przemyśleć, pod opieką lekarza prowadzącego oraz dietetyka, dietę eliminującą dany alergen.
Według dostępnej wiedzy medycznej brak jest podstaw do stosowania przez kobiety ciężarne eliminacji w celu zapobiegania wystąpienia u dzieci problemów alergicznych. Wycofanie potencjalnie alergizujących pokarmów nie ma wpływu na mniejszą częstość alergii u potomstwa. Niewskazane jest również, aby profilaktycznie zalecać dietę eliminacyjną matkom karmiącym. Wiemy, że w mleku kobiecym są alergeny pokarmowe pochodzące z diety matki, ale występują one w dużo niższym stężeniu. Dodatkowo w mleku kobiecym znajduje się ponad 2600 białek – bioaktywne molekuły, które można traktować jako immunoterapię i trening dla układu immunologicznego dziecka.
Temat alergii pokarmowych jest dość powszechny i niestety dotyka coraz większej liczby dzieci.
Alergia na białko mleka krowiego
Alergia na białko mleka krowiego jest najczęściej i najwcześniej występującą alergią pokarmową u dzieci. W przypadku reakcji niezależnej od przeciwciał IgE, mimo dotkliwych objawów, pojawiają się problemy diagnostyczne. Im późniejsza diagnoza i eliminacja tym większe ryzyko osłabienia nabłonka jelitowego, a to prowadzi do rozwoju reakcji alergicznej. U większości dzieci tolerancja na mleko krowie pojawia się około 5. roku życia, ale odnotowujemy odsetek pacjentów, u których objawy alergii nie ustępują i mogą się utrzymywać przez całe życie.
Przy alergii na białko mleka krowiego (zależnej od przeciwciał IgE) reakcje obserwujemynie tylko mleko krowie, ale również mleko kozie, bawole, owcze oraz końskie.
Należy zwrócić uwagę na produkty zawierające białko mleka krowiego lub innego pochodzenia:
oraz na takie, które mogą zawierać białko mleka krowiego lub innego pochodzenia:
czekolada, tłuszcze roślinne do smarowania pieczywa, wypieki, lody, nugat, desery, zabielacz do kawy, margaryna, puree ziemniaczane.
Mleko jest źródłem białka, tłuszczu, wapnia, fosforu, witamin A, D, E, K oraz wit. z grupy B.
Zawartość białka w mleku krowim (obejmującym ponad 20 protein) wynosi od 2,5 do 4,2% i jest około 1,5-2-krotnie wyższa niż w mleku ludzkim. β-laktoglobulina (brak jej w mleku kobiecym) i kazeina to podstawowe alergeny występujące w mleku krowim.
Laktoza to cukier występujący w mleku a nietolerancja laktozy związana jest z brakiem enzymu laktazy – nie należy tego mylić z alergią!
W przypadku nietolerancji laktozy wystarczy wprowadzić produkty oznaczone przez producenta jako bez laktozy.
Napoje roślinne – alternatywa dla mleka krowiego?
Obserwujemy tendencję zastępowania mleka odzwierzęcego, napojami roślinnymi. W 2007 roku przepisy szczegółowo określiły, że termin mleko jest zarezerwowane tylko i wyłącznie dla wydzieliny wymion ssaków.
Napoje roślinne powstające na drodze wodnej ekstrakcji nasion oleistych oraz orzechów nie mogą być nazywane mlekiem, aby nie wprowadzać konsumenta w błąd.
Na rynku dostępne są napoje roślinne otrzymywane:
z nasion roślin strączkowych (sojowe),
ze zbóż (ryżowe, owsiane, jaglane, orkiszowe, gryczane),
pestek lub orzechów (migdałowe, z orzechów laskowych),
kokosa,
konopi.
W obiegowej opinii napoje roślinne są traktowane jako zamiennik mleka.
Niezależnie od surowca wartość odżywcza mleka odzwierzęcego będzie inna niż napoju roślinnego. Co ważne, niemożliwe jest również przeniesienie wartości odżywczej z surowca roślinnego na gotowy produkt. Procentowy udział surowca (orzecha, zboża, pestek) ostatecznie w napoju roślinnym dochodzi średnio do 6%, resztę stanowi woda. Wartość odżywcza takiego produktu będzie dużo niższa.
Dodatkowo należy wybierać napoje roślinne fortyfikowane, wzbogacane o wapń, wit. D, żelazo, cynk czy wit. B12.
Warto czytać dokładnie etykiety, ponieważ cześć napojów roślinnych może zawierać dodatek zagęstników, stabilizatorów i emulgatorów oraz cukier, fruktozę, maltodekstrynę, sól.
Według Zasad żywienia zdrowych niemowląt Stanowiska Polskiego Towarzystwa Gastroenterologii, Hepatologii i Żywienia Dzieci z 2021 roku bezmleczne napoje roślinne nie pokrywają podstawowego zapotrzebowania dziecka w 1. roku życia na składniki odżywcze. Nie mogą stanowić alternatywy dla preparatów mlekozastępczych.
Przegląd napojów roślinnych
Napój sojowy – pod względem zawartości białka najbardziej podobny do mleka. Białko sojowe może wywoływać reakcje alergiczne (często współtowarzysząca jest alergia na białko mleka i białko soi). Kontrowersyjnymi składnikami produktów sojowych są także fitoestrogeny, które mogą powodować zachwianie równowagi hormonalnej. Z drugiej strony fitoestrogeny mają korzystny wpływ na łagodzenie skutków przemian hormonalnych u kobiet w okresie menopauzy. Plusem napoju sojowego jest zawartość lecytyny (fosfolipid), która ma istotne znaczenie dla funkcjonowania układu neurologicznego.
Napój ryżowy – ziarno ryżu jest bogate w skrobię i zawiera najwięcej węglowodanów wśród napojów roślinnych – prawie trzy razy więcej w porównaniu do mleka krowiego. Zawartość białka około 1% – niewskazany dla najmłodszych ze względu na kumulację arsenu.
Napój z owsa – zawiera błonniki- ß-glukany, które mają właściwości immunomodulujące, przeciwzapalne, przeciwnowotworowe i prebiotyczne tzn. stymuluje rozwój prawidłowego mikrobiomu jelit, a także poprawia perystaltykę jelit. Owies zawiera żelazo, potas, wapń, magnez, cynk i selen oraz witaminy z grupy B. Białko owsa cechuje się zawartością aminokwasów egzogennych.
Napój kokosowy (to nie to samo co woda kokosowa) – jest bogaty w błonnik, witaminę E i z grupy B, żelazo, selen, wapń, sód, magnez i fosfor. Zawiera średniołańcuchowe nasycone kwasy tłuszczowe, w tym kwas laurynowy. Napój cechuje niska zawartością cukru i kremową konsystencją – dlatego jest dobrą alternatywą dla śmietanki.
Napój migdałowy – wartość napoju wiąże się z tym, że migdały mają wysoką zawartość tłuszczów jednonienasyconych, małą kwasów nasyconych, istotną zawartością witaminy E, magnezu, potasu i łatwo dostępnego wapnia.
Napój z konopi – ma charakterystyczny orzechowy smak i jest bogaty w kwasy omega-3, zawiera fitosterole, witaminę C, wit. z grupy B, beta-karoten, wapń, żelazo, potas, fosfor oraz 10 egzogennych aminokwasów, czyli takich, których organizm nie jest w stanie sam wytworzyć.
Czym zastąpić produkty mleczne na diecie eliminacyjnej?
Warto pamiętać, że smak sera uzyskamy, używając płatki drożdżowe lub tempeh, czyli przefermentowaną soję.
Twarożek można przygotować z orzechów nerkowca.
Śmietanę zastąpimy mleczkiem kokosowym lub śmietaną sojową.
Na rynku znajdziemy jogurty roślinne z kokosa, nerkowca czy migdałów.
Dobrą alternatywą dla tradycyjnych lodów są mrożone owoce zblendowane z napojem kokosowym lub roślinnym jogurtem.
Produkty bogate w wapń, o które warto zadbać na diecie bez mleka to figi, sezam, pasta tahini, brokuły, pomarańcze, migdały i fasola.
Nieodpowiednio prowadzona dieta eliminacyjna może prowadzić do powikłań pod postacią niedoboru masy ciała i wzrostu, niedożywienia, hipowitaminozy.
Sama masa ciała nie świadczy o odżywieniu. Odnotowujemy wiele przypadków pacjentów z nadwagą/otyłością z równoczesnym niedożywieniem jakościowym.
Każde restrykcje powinny być wprowadzane na podstawie wyników badań w zestawieniu z objawami. Zawsze warto zasięgnąć porady u lekarza oraz dietetyka, który pomoże prawidłowo zbilansować dietę.
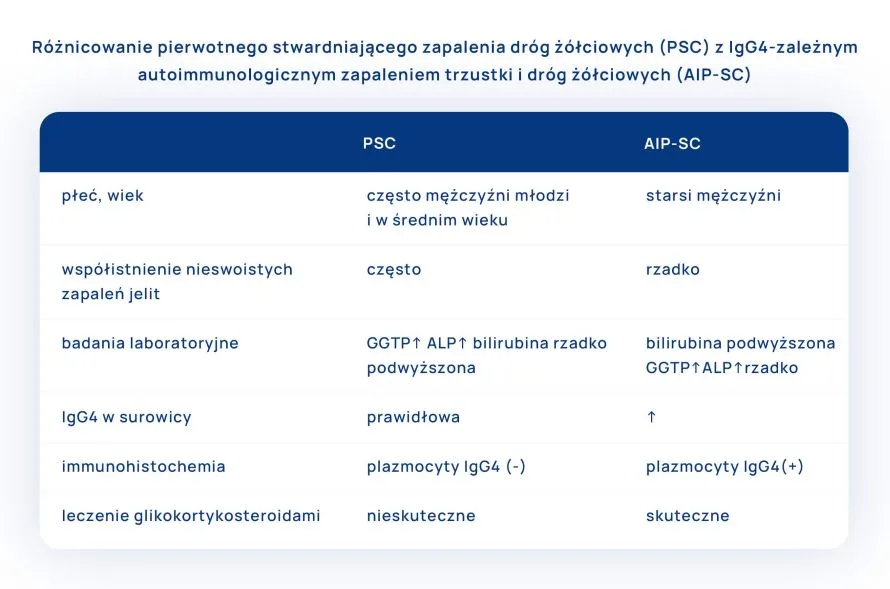
Pierwotne stwardniające zapalenie dróg żółciowych – PSC (primary sclerosing cholangitis) jest przewlekłą, cholestatyczną chorobą, która ma charakter postępujący. Charakteryzuje się naciekami zapalnymi i postępującym zwłóknieniem zewnątrzwątrobowych i wewnątrzwątrobowych dróg żółciowych i pęcherzyka żółciowego. Choroba jest nieodwracalna, u pacjentów często występuje bezobjawowa cholestaza, ale u wielu z czasem rozwijają się postępujące zwężenia dróg żółciowych, prowadzące do nawracającego zapalenia dróg żółciowych, marskości żółciowej i schyłkowej niewydolności wątroby.
Pierwotne stwardniające zapalenie dróg żółciowych (PSC) jest klasyczną manifestacją wątrobowo-żółciową choroby zapalnej jelit. Z PSC bardzo często współistnieją nieswoiste zapalenia jelit, a szczególnie wrzodziejące zapalenie jelita grubego (WZJG). Leczenie farmakologiczne nie spowalnia postępu choroby, a wielu pacjentów wymaga przeszczepienia wątroby, po którym istnieje ryzyko nawrotu choroby. Szczególny niepokój budzi zwiększona częstość występowania raka wątroby i dróg żółciowych, która nie jest związana z ciężkością zwłóknienia dróg żółciowych. Ryzyko raka jelita grubego jest również zwiększone u pacjentów ze współistniejącą chorobą zapalną jelit.
Rozpoznanie pierwotnego stwardniającego zapalenia dróg żółciowych (PSC) opiera się na wynikach klinicznych, laboratoryjnych, obrazowych i histologicznych.
Kto choruje najczęściej?
Choroba może rozwinąć się w każdym wieku, ale najczęściej dotyczy mężczyzn w wieku od 20. do 40. roku życia (mężczyźni chorują dwukrotnie częściej niż kobiety). Typowy chory z pierwotnym stwardniającym zapaleniem dróg żółciowych (PSC) to młody lub w średnim wieku mężczyzna z objawami i nieprawidłowymi wynikami badań laboratoryjnych (wskaźnikami cholestazy) oraz z nieswoistym zapaleniem jelita grubego. Średni czas przeżycia chorych z PSC bez przeszczepiania wątroby wynosi 12–17 lat.
Mechanizm powstawania choroby
Mechanizmy powstawania pierwotnego stwardniającego zapalenia dróg żółciowych (PSC) nie jest w pełni poznany i prawdopodobnie ma bardzo złożony charakter. Dużą rolę przypisuje się zmienionej odpowiedzi immunologicznej na czynniki infekcyjne, toksyczne, a nawet środowiskowe, która prowadzi do nieprawidłowej aktywacji układu odpornościowego i utrwalenia procesu zapalnego. Za tą hipotezą przemawia częste współistnienie PSC z chorobami autoimmunizacyjnymi takich jak: wrzodziejące zapalenie jelita grubego, autoimmunologiczne zapalenie trzustki, toczeń układowy trzewny, cukrzyca, zapalenia naczyń, autoimmunologiczne choroby tarczycy, zapalenie pęcherzyków płucnych, łuszczyca, RZS, celiakia, sarkoidoza, idiopatyczna plamica małopłytkowa stwardnienie rozsiane, zespół Sjögrena, zapalenie wielomięśniowe, bielactwo – ok. 25% pacjentów z pierwotnym stwardniającym zapaleniem dróg żółciowych (PSC) ma jedną z wymienionych chorób. Na tło immunologiczne może również wskazywać obecność autoprzeciwciał. Trzeba jednak podkreślić, że objawy typowe dla zaburzeń immunologicznych są mniej wyraźne w PSC w porównaniu z innymi chorobami wątroby o autoimmunologicznej patogenezie.
Badania wykazały, że również czynniki genetyczne np. obecność haplotypu HLA B8 i DR3 oraz polimorfizmy genów zlokalizowanych w sąsiedztwie HLA, kodujące między innymi czynnik martwicy nowotworów TNF (tumor necrosis factor) i niektóre białka dopełniacza (również uczestniczące w procesach immunologicznych) mają istotny wpływ na ryzyko wystąpienia choroby i łączą we wspólny mechanizm patogenezy pierwotnego stwardniającego zapalenia dróg żółciowych (PSC) czynniki genetyczne i immunologiczne. Częste współistnienie wrzodziejącego zapalenia jelita grubego może wskazywać na uszkodzenia dróg żółciowych przez bakterie, toksyny bakteryjne i reakcje immunologiczne spowodowane przez nieprawidłowe cząsteczki adhezyjne i limfocyty T przemieszczające się krążeniem wrotnym z chorego jelita do wątroby.
Objawy pierwotnego stwardniającego zapalenia dróg żółciowych
U 15-45% pacjentów choroba ma przebieg bezobjawowy (diagnostyka podejmowana jest wtedy z powodu przypadkowo stwierdzonych, nieprawidłowych wyników badań laboratoryjnych).
Objawem charakterystycznym, ale niewystępującym u wszystkich chorych jest świąd skóry, który obserwuje się u 25–59% chorych z PSC w chwili rozpoznania. Natomiast najczęstszym objawem występującym u ponad 65% chorych jest uczucie przewlekłego zmęczenia, nieproporcjonalnego do obciążeń psychofizycznych pacjenta.
Dość często (16–37%), szczególnie u chorych z „dominującym zwężeniem dróg żółciowych”, pojawiają się objawy obserwowane w kamicy dróg żółciowych: bóle w prawym podżebrzu, znaczne, przemijające zażółcenie skóry i błon śluzowych, przeczosy, a nawet typowe objawy zapalenia dróg żółciowych (5–28%). W stadium zaawansowanym choroby pojawia się coraz wyraźniejsza żółtaczka, spadek masy ciała, powiększenie wątroby oraz inne objawy niewydolności wątroby lub nadciśnienia wrotnego (wodobrzusze, żylaki przełyku) oraz powiększenie śledziony.
Diagnostyka laboratoryjna PSC
U chorych z pierwotnym stwardniającym zapaleniem dróg żółciowych(PSC) w badaniach laboratoryjnych stwierdza się obecność cholestazy:
obserwuje się umiarkowany wzrost poziomu parametrów wątrobowych – ALT (aminotransferazy alaninowej) i AST (aminotransferazy asparaginowej) 2-4 razy przekroczona górna granicy wartości referencyjnych,
hipergammaglobulinemię IgG i IgM (45-80% chorych),
obserwuje się obecność autoprzeciwciał.
Oznaczanie autoprzeciwciał występujących w pierwotnym stwardniającym zapaleniu dróg żółciowych (PSC) nie ma istotnego znaczenie w rozpoznaniu choroby. Bywa przydatne w różnicowaniu zespołów nakładania lub rozpoznawaniu współistniejących z PSC innych chorób autoimmunologicznych.
Zmiany w drogach żółciowych mogą również elementem chorób o różnej naturze. Różnicowanie zmian wtórnych w drogach żółciowych z PSC jest trudne. Wówczas, w różnicowaniu pomocne będą badania autoprzeciwciał, wywiady co do współistniejących chorób autoimmunizacyjnych, a przede wszystkim badanie kolonoskopowe i badanie kalprotektyny w kale.
Najczęstsze, poza PSC, przyczyny zmian patologicznych w drogach żółciowych
Wtórne stwardniające zapalenie dróg żółciowych:
kamica dróg żółciowych,
zakażenia,
zapalenie trzustki,
zabiegi chirurgiczne na drogach żółciowych,
urazy jamy brzusznej,
polekowe,
chemoembolizacja zmian nowotworowych przez tętnicę wątrobową.
Zmiany w przebiegu innych chorób dróg żółciowych podobne do zmian w PSC:
choroba IgG4-zależna,
eozynofilowe zapalenie,
cholangiopatia w przebiegu AIDS,
niedokrwienne zapalenie,
wady wrodzone,
rak dróg żółciowych.
Szczególnej uwagi wymaga różnicowanie pierwotnego stwardniającego zapalenia dróg żółciowych(PSC) z najczęstszą formą choroby IgG4-zależnej, jaką jest autoimmunologiczne zapalenie trzustki i dróg żółciowych (AIP-SC, IgG4 associated autoimmune pancreatitis-sclerosing cholangitis).
Całkowite wyleczenie pierwotnego stwardniającego zapalenia dróg żółciowych PSC jest niemożliwe. Stosowane leczenie farmakologiczne jedynie zmniejsza nasilenie objawów choroby oraz poprawia parametry laboratoryjne. Postępowanie endoskopowe wykonuje się w przypadku narastania objawów, nie powoduje jednak ustąpienia choroby. Leczenie farmakologiczne w PSC jest długotrwałe. Podczas leczenia należy również zwrócić uwagę na powikłania choroby, do których należy między innymi niedobór witamin rozpuszczalnych w tłuszczach (A, D3, E) i wapnia.
Piśmiennictwo
Interna Szczeklika, Wyd. Medycyna Praktyczna Kraków 2021.
Ferri P, Simões E Silva A, Campos Silva S, de Aquino D, Fagundes E, Marques de Miranda D, Ferreira A.:The Role of Genetic and Immune Factors for the Pathogenesis of Primary Sclerosing Cholangitis in Childhood. Gastroenterol Res Pract. 2016.
Y. Shoenfeld, P.L Meroni: The general practice guide to autoimmune disease, Pabst science publishers Lengerich 2012.
K. Simon red.: Diagnostyka chorób wątroby, Termedia Wyd.Medyczne, Poznań 2012.
Biotyna (witamina H, witamina B7) to witamina, która przez bardzo dobrze kojarzone działanie poprawiające kondycję włosów, skóry i paznokci jest częstym wyborem zwłaszcza Pań odwiedzających aptekę. Czy jednak mamy świadomość, że nawet preparat na wzmocnienie włosów dostępny bez recepty może znacząco wpłynąć na nasze wyniki badań?
Czym jest biotyna?
Biotyna to heterocykliczny związek chemiczny, zaliczany do grupy witamin. Nazywana inaczej witaminą H lub B7, nie jest syntetyzowana w organizmie człowieka. Podstawowym jej źródłem jest zatem dieta oraz w niewielkim stopniu synteza przez bakterie jelitowe w jelicie grubym.
Najbogatszymi w biotynę produktami spożywczymi są: żółtko jaja kurzego, wątróbka, orzechy włoskie, rośliny strączkowe, produkty mleczne (mleko krowie, sery), drożdże piwne, ryby takie jak sardynki czy łosoś.
Biotyna (witamina H, witamina B7) – działanie
Najbardziej kojarzonym działaniem biotyny jest jej korzystne działanie na stan skóry, paznokci oraz wpływający na ilość i jakość włosów. Warto jednak zaznaczyć, że działanie to nie znalazło jednoznacznego potwierdzenia w badaniach naukowych.
Zalecane dzienne spożycie biotyny dla osób dorosłych wynosi ok. 30 µg/dzień. Szacuje się, że dzienne średnie spożycie biotyny wśród osób dorosłych mieści się w przedziale 30 a 70 µg na dzień, co oznacza, że w przypadku prawidłowo zbilansowanej diety zdrowej dorosłej osoby prawdopodobieństwo wystąpienia niedoboru jest niskie.
Biotyna (witamina H, witamina B7) – niedobór
Niedobór biotyny może być nabyty lub wrodzony. Niedobór nabyty może wiązać się z dużą zawartością awidyny w diecie, związku występującego np. w surowym białku jaja kurzego (obróbka termiczna neutralizuje jego działanie). Niski poziom biotyny obserwuje się również u pacjentów z nieswoistym zapaleniem jelit, łojotokowym zapaleniem skóry czy w chorobie Leinera. Również pacjenci żywieni pozajelitowo są narażenie na niedobory witaminy H.
Pacjenci stosujący leki przeciwdrgawkowe jak np. fenobarbital, kwas walproinowy, karbamazepiny, prymidon, fenytoina, a także leki sterydowe mogą być szczególnie narażeni na niedobór biotyny.
Alkohol, izotretynoina oraz antybiotyki poprzez wpływ na mikrobiotę jelitową mogą powodować zaburzenia wchłaniania biotyny i tym samym prowadzić do jej niedoborów.
Wrodzony niedobór biotyny spowodowany jest autosomalną cechą recesywną prowadzącą do braku enzymów biorących udział w metabolizmie biotyny.
W przypadku niedoboru biotyny obserwować można wypadanie włosów, łamliwość i kruchość paznokci, łojotokowe zapalenie skóry, wypryskowe wysypki skórne, zmęczenie, wzmożoną senność, bóle mięśniowe, zapalenie spojówek, spadek odporności organizmu.
Biotyna (witamina H, witamina B7) a wyniki badań laboratoryjnych
Diagnostyka większości chorób opiera się na dwóch składowych: obserwacji stanu klinicznego, czyli występowaniu objawów charakterystycznych dla danego schorzenia oraz na wykonaniu badań laboratoryjnych. Jedną z najpopularniejszych metod wykonywania oznaczeń stężeń wybranego parametru w surowicy są metody immunochemiczne.
Immunochemiczne metody badań laboratoryjnych polegają na mieszaniu badanego materiału z roztworem zawierającym znakowane biotyną przeciwciała. Są one charakterystyczne dla oznaczanych w próbce substancji (antygenów). W zależności od typu badania powstają połączenia przeciwciało-antygen-przeciwciało (metoda niekompetycyjna) lub antygen-przeciwciało (metoda kompetycyjna). Tak utworzone kompleksy łączą się z podłożem za pośrednictwem cząsteczki biotyny w przeciwciele. Oznaczenia stężenia badanego antygenu dokonuje się na podstawie ilości połączonych z podłożem kompleksów. Na tym etapie cały wynik badania może zaburzyć „egzogenna”, czyli suplementowana biotyna (która, łącząc się z podłożem, uniemożliwi połączenie się z nim badanego antygenu).
Przykłady badań, które ulegają zafałszowaniu przy jednoczesnej suplementacji biotyną
Optymalny odstęp między suplementacją a wykonaniem badań
Minimalny odstęp czasu między stosowaniem małych dawek biotyny a badaniami laboratoryjnymi to 1 doba, choć w wielu przypadkach uznaje się, że jest to dostęp za mały i sugeruje utrzymanie co najmniej 48 h przerwy między zażyciem preparatu a wykonaniem badań. W przypadku wyższych dawek biotyny (wyższych niż 5mg/dobę) minimalny odstęp to 3-7 dni. W badaniach podkreśla się, że w tym przypadku zalecany odstęp może być sprawą bardzo indywidualną zależną od cech osobniczych i stosowanej dawki.
Piśmiennictwo
Patel DP, Swink SM, Castelo-Soccio L. A Review of the Use of Biotin for Hair Loss. Skin Appendage Disord. 2017; 3(3): 166–169.
Said HM. Biotin: biochemical, physiological and clinical aspects. Subcell Biochem. 2012; 56: 1–19.
León-Del-Río A. Biotin in metabolism, gene expression, and human disease. J Inherit Metab Dis. 2019; 42(4): 647–654
Thompson KG, Kim N. Dietary supplements in dermatology: A review of the evidence for zinc, biotin, vitamin D, nicotinamide, and Polypodium. J Am Acad Dermatol. 2021; 84(4): 10421050.
Ostrowska M., Bartoszewicz Z., Bednarczuk T., Walczak K., Zgliczyński W., Glinicki Z.: The effect of biotin interference on the results of blood hormone assays. Endokrynologia Polska, 2019, vol. 70, no. 1/2019, ISSN 0423–104X.
Piketty ML, Prie D, Sedel F, et al.: High-dose biotin therapy leading to false biochemical endocrine profiles: validation of a simple method to overcome biotin interference. Clin Chem Lab Med. 2017; 55(6): 817–825.
Piketty ML, Polak M, Flechtner I, et al. False biochemical diagnosis of hyperthyroidism in streptavidin-biotin-based immunoassays: the problem of biotin intake and related interferences. Clin Chem Lab Med. 2017; 55(6): 780–788.
Evans E, Piccio L, Cross AH. Use of Vitamins and dietary supplements by patients with multiple sclerosis: A review. JAMA Neurol. 2018; 75(8): 1013–1021.
Poniższy artykuł jest zapisem webinaru, który odbył się 17.05.2023 r.
Stres oksydacyjny to stan zaburzenia równowagi między reaktywnymi formami tlenu, czyli wolnymi rodnikami, a zdolnością ciała do ich eliminacji. Oksydanty, posiadające jeden wolny elektron, mogą uszkodzić komórki, białka, tłuszcze i DNA. Chociaż wolne rodniki powstają naturalnie podczas oddychania, istnieją też inne sytuacje sprzyjające ich produkcji. W tym artykule zostanie omówiony ten proces powstawania i sposoby ochrony przed nadmiernym gromadzeniem się oksydantów w organizmie.
Stres oksydacyjny a stres nitrozacyjny
Stres oksydacyjny jest zjawiskiem dobrze znanym w medycynie. Polega on na produkcji reaktywnych form tlenu, nazywanych oksydantami. Jednak równie ważnym, choć mniej rozpowszechnionym mechanizmem, jest stres nitrozacyjny. W tym przypadku w organizmie produkowane są reaktywne formy azotu w procesie nitronizacji. Niestety, te substancje mogą powodować zmiany w strukturze białek, które następnie nie funkcjonują prawidłowo.
Chociaż stres nitrozacyjny nie jest tak często omawiany, jak stres oksydacyjny, oba te mechanizmy są ze sobą ściśle powiązane. Nadmiar wolnych rodników, wynikający ze stresu oksydacyjnego, prowadzi do spadku produkcji energii w mitochondriach, naszych komórkowych centrach energetycznych. To z kolei może prowadzić do różnych dolegliwości, co podkreśla znaczenie monitorowania obu tych procesów podczas diagnozowania pacjentów.
System andyoksydacyjny a geny
Współczesne badania wskazują, że brak równowagi w stresie oksydacyjnym może mieć swoje korzenie w genach oraz w mechanizmach regulujących ekspresję genów.
Kluczowym elementem w tym kontekście jest czynnik transkrypcyjny o nazwie Nrf2. Chociaż specjaliści od genetyki mogliby zagłębić się w ten temat bardziej szczegółowo, warto podkreślić, że Nrf2 jest uważany za główny regulator odpowiedzi oksydacyjnej. Jego działanie nie ogranicza się jedynie do enzymów antyoksydacyjnych. W rzeczywistości moduluje on ekspresję setek genów, w tym tych odpowiedzialnych za różnorodne procesy, takie jak przebudowa tkanek, rakotwórczość, dysfunkcje poznawcze czy nawet zachowania uzależniające. Ta złożoność działania Nrf2 pokazuje, jak ważne jest zrozumienie roli genetyki w kontekście stresu oksydacyjnego i systemu antyoksydacyjnego.
Grupy ryzyka wystąpienia stresu oksydacyjnego
Stres oksydacyjny to stan, który może dotknąć każdego z nas w różnych okolicznościach, takich jak urazy czy zabiegi operacyjne. Jednak istnieją pewne grupy osób, które są na niego szczególnie narażone:
Właściciele dużych firm i menadżerowie: Ci ludzie często pracują pod presją, mają wiele obowiązków i są narażeni na chroniczny stres, co może prowadzić do wzmożonej produkcji wolnych rodników.
Sportowcy wyczynowi: Pomimo korzyści zdrowotnych płynących z regularnej aktywności fizycznej, nadmierny wysiłek fizyczny może prowadzić do produkcji wolnych rodników. Wysiłek fizyczny o charakterze umiarkowanym jest korzystny, ale jego nadmiar może prowadzić do problemów zdrowotnych, takich jak dysregulacja układu immunologicznego.
Młodzież: Współczesna młodzież często boryka się z wieloma wyzwaniami, takimi jak depresja, zaburzenia snu, niewłaściwe odżywianie czy nadmiar obowiązków. Brak snu, niewłaściwa dieta i brak aktywności fizycznej sprawiają, że są oni szczególnie narażeni na stres oksydacyjny.
Warto zwrócić uwagę, że stres oksydacyjny i stres emocjonalny to dwa różne pojęcia, ale oba mogą wpływać na zdrowie. W obu przypadkach kluczem jest zrozumienie, jakie czynniki mogą prowadzić do stresu oksydacyjnego i jak można go unikać lub minimalizować jego skutki.
Przyczyny powstawania stresu oksydacyjnego
Stres oksydacyjny to stan, który może dotknąć każdego z nas, ale istnieją pewne czynniki, które zwiększają ryzyko jego wystąpienia:
Przewlekły stres: Krótkotrwały stres może być mobilizujący, ale przewlekły stres prowadzi do produkcji prozapalnych cytokin, takich jak cytokina nr 6.
Nieodpowiednia dieta: Spożywanie tłuszczów trans, produktów przetworzonych, wędzonych, spleśniałych czy smażonych w głębokim tłuszczu może prowadzić do stresu oksydacyjnego.
Używki: Palenie papierosów, nadmierne spożywanie alkoholu czy używanie niektórych leków, takich jak antykoncepcyjne, antydepresanty, sterydy czy antykoagulanty.
Zaburzenia snu: Zbyt mała ilość snu czy zaburzenia cyklu dobowego.
Choroby układowe: Osoby z chorobami układowymi są bardziej narażone na stres oksydacyjny.
Brak aktywności fizycznej lub nadmierna, cykliczna aktywność fizyczna.
Nadmierna ekspozycja na słońce bez odpowiedniej ochrony.
Wiek – z wiekiem nasze mechanizmy obronne przed wolnymi rodnikami ulegają degradacji.
Rozumienie tych czynników jest kluczem do zapobiegania stresowi oksydacyjnemu i dbania o zdrowie na poziomie komórkowym.
Pierwsze symptomy stresu oksydacyjnego
Stres oksydacyjny, chociaż może wydawać się abstrakcyjnym terminem, ma bardzo konkretne objawy, które mogą wpływać na nasze codzienne życie. Warto zwrócić uwagę na pierwsze symptomy, które mogą wskazywać na obecność stresu oksydacyjnego w organizmie:
problemy z koncentracją,
osłabienie i przewlekłe zmęczenie,
bóle głowy,
problemy z trawieniem, takie jak zaparcia,
wysypki skórne,
bóle stawów,
utrata siły mięśniowej.
Chociaż te objawy są nieswoiste i mogą występować w wielu innych sytuacjach klinicznych, ich obecność, zwłaszcza w połączeniu, powinna skłonić nas do refleksji. Jeśli doświadczamy kilku z tych objawów jednocześnie, warto zastanowić się nad tym, czy nie są one spowodowane właśnie przez stres oksydacyjny. Wiele osób może doświadczać tych objawów od czasu do czasu, ale jeśli stają się one przewlekłe i utrzymują się przez dłuższy czas, mogą wskazywać na poważniejsze problemy zdrowotne. Długotrwały stres oksydacyjny, jeśli nie zostanie rozpoznany i odpowiednio leczony, może prowadzić do wielu chorób przewlekłych i poważnych komplikacji zdrowotnych. Dlatego tak ważne jest, aby nie lekceważyć tych objawów i podjąć odpowiednie kroki w celu ich rozpoznania i leczenia.
Choroby powiązane ze stresem oksydacyjnym
Stres oksydacyjny, choć nie jest zjawiskiem nowym, stał się w ostatnich latach przedmiotem intensywnych badań. Współczesna nauka coraz bardziej dostrzega jego wpływ na wiele jednostek chorobowych. Niektóre z chorób i schorzeń, które mogą być powiązane ze stresem oksydacyjnym to:
Współistnienie wielu z tych chorób z podwyższonym poziomem stresu oksydacyjnego wskazuje na potrzebę głębszego zrozumienia tego zjawiska i jego wpływu na zdrowie ludzi. Kluczem jest nie tylko identyfikacja stresu oksydacyjnego, ale także zrozumienie, jak nasz organizm broni się przed nim i jakie mechanizmy obronne są w nim aktywowane w odpowiedzi na nadmiar wolnych rodników.
System antyoksydacyjny – obrona przed stresem oksydacyjnym
Organizm posiada system antyoksydacyjny, który składa się z enzymów i przeciwutleniaczy, chroniących przed uszkodzeniami oksydacyjnymi.
Enzymy antyoksydacyjne to:
dysmutaza ponadtlenkowa, która neutralizuje ponadtlenki,
katalaza, która rozkłada nadtlenek wodoru,
peroksydaza glutationowa, która współdziała z glutationem.
Aby system działał efektywnie, ważne jest dostarczanie organizmowi niezbędnych składników odżywczych.
Diagnostyka stresu oksydacyjnego
Diagnostyka stresu oksydacyjnego opiera się na indywidualnym podejściu do pacjenta. Współczesne narzędzia diagnostyczne skupiają się na analizie różnych biomarkerów, które wskazują na obecność stresu oksydacyjnego i nitronizacyjnego w organizmie. Ważne jest również uwzględnienie elementów przeciwutleniających i proutleniających, które pokazują, jak organizm radzi sobie z wolnymi rodnikami. W przyszłości kluczowymi elementami w diagnozowaniu stresu oksydacyjnego mogą się stać badania genetyczne oraz analiza przeciwciał.
W Polsce w diagnostyce stresu oksydacyjnego bada się poziom glutationu oraz długość telomerów, potocznie przez pacjentów nazywaną „wiekiem biologicznym”. Inne testy to analiza 8-hydroksy-2-deoksyguanozyny, wskaźnika oksydacyjnego uszkodzenia DNA, oraz aktywność enzymów antyoksydacyjnych, takich jak dysmutaza ponadtlenkowa.
Ważne są również badania poziomu peroksydacji lipidów, markerów stanu zapalnego oraz niedoborów antyoksydantów, w tym witaminy C. Dodatkowo, oznaczanie przeciwciał autoimmunizacyjnych oraz poziomu koenzymu Q10 dostarcza informacji o stanie zdrowia pacjenta w kontekście stresu oksydacyjnego.
Dobre efekty przynosi też oznaczanie poziomu pierwiastków i metali ciężkich w erytrocytach, choć w Polsce ta forma diagnostyki jest stosowana rzadziej. Istotne jest także badanie neurotransmiterów w kontekście cyklu dobowego, zwłaszcza u pacjentów z depresją. Kluczem do skutecznej diagnostyki jest indywidualne podejście do pacjenta oraz dokładny wywiad medyczny.
Glutation
Jednym z najważniejszych antyoksydantów w organizmie ludzkim jest glutation. W zdrowych komórkach stosunek glutationu całkowitego do zredukowanego powinien wynosić 0,99. Wartości poniżej tej normy wskazują na obecność stresu oksydacyjnego, a wartość 0,63 wskazuje na poważne zaburzenia.
Glutation nie tylko redukuje wolne rodniki, ale także współpracuje z witaminą C w szlaku glutationowo-askorbinowym, który jest kluczowym elementem systemu antyoksydacyjnego. Dzięki glutationowi witamina C może się odnawiać. Bez glutationu komórki ulegają utlenianiu, a odporność organizmu ulega osłabieniu. Każda komórka w organizmie produkuje glutation, ale potrzebuje do tego odpowiednich substratów. Glutation jest niezbędny do syntezy i naprawy DNA, a jego brak może prowadzić do uszkodzenia DNA.
Odpowiednie poziomy glutationu pomogą w walce z miażdżycą, opóźniając produkcję, tworzenie płytek w tętnicach. Bez glutationu żaden antyoksydant nie może odpowiednio spełniać swojej funkcji, gdyż nie będzie się odnawiać, czyli nie będzie chronić przed różnego rodzaju chorobami.
Suplementacja glutationu
Chociaż glutation jest dostępny w formie suplementów, takich jak tabletki czy formy liposomalne, jego suplementacja w tej formie jest obecnie uważana za nieskuteczną.
Glutation zawarty w produktach spożywczych, takich jak awokado, szparagi czy brokuły, rozkłada się w jelicie cienkim na trzy aminokwasy. Kluczowym aminokwasem jest L-cysteina, z której organizm może wytwarzać glutation. Jednakże, aby produkować L-cysteinę, potrzebujemy metioniny, aminokwasu egzogennego, który musi być dostarczany w diecie.
Osoby na restrykcyjnych dietach, takich jak dieta wegetariańska, mogą mieć niski poziom metioniny, ale można to zrównoważyć spożywając odpowiednie produkty, takie jak sezam czy orzechy brazylijskie. Ważne jest, aby podejść do suplementacji glutationu z odpowiednią wiedzą dietetyczną, aby była ona skuteczna.
Badanie długości telomerów
Informacji o występowaniu stresu oksydacyjnego w organizmie może też dostarczyć badanie długości telomerów. Telomery są końcowymi fragmentami chromosomów, które skracają się w miarę starzenia się komórek. Przedwczesne skracanie telomerów może wskazywać na obecność stresu oksydacyjnego.
W naturalny sposób telomery skracają się z wiekiem, ale styl życia człowieka, takie czynniki jak palenie papierosów, otyłość czy brak aktywności fizycznej, mogą przyspieszać ten proces. Aby zachować długość telomerów i opóźnić proces starzenia, ważne jest prowadzenie zdrowego stylu życia, unikanie czynników stresogennych i dbanie o odpowiednią dietę oraz aktywność fizyczną.
Istotnym czynnikiem w kontekście długości telomerów jest działanie tak zwanej telomerazy, czyli enzymu, który wydłuża telomery jeszcze przed replikacją, w intensywnie dzielących się komórkach. Aktywna telomeraza wpływa na regenerację komórek w telekamerach.
Interpretacja badania długości telomerów
Długość telomerów jest kluczowym wskaźnikiem zdrowia komórkowego i może być używana jako wskaźnik biologicznego wieku organizmu. Chociaż naturalnie skracają się one z wiekiem, wiele czynników może wpływać na ich długość, zarówno pozytywnie, jak i negatywnie.
Wyniki badań sugerują, że doświadczenia i styl życia matki przed i w trakcie ciąży mogą wpływać na długość telomerów jej dziecka. Wsparcie społeczne, jakie matka otrzymuje w dzieciństwie, może wpływać na zdrowie jej przyszłego dziecka na poziomie komórkowym. Z drugiej strony, negatywne czynniki, takie jak otyłość czy palenie papierosów, mogą skracać telomery, zwiększając ryzyko różnych chorób u potomstwa.
Na długość telomerów jej dziecka, zwłaszcza u płci męskiej, może też wpływać stres psychiczny matki podczas ciąży. Niezależnie od płci dziecka na skracanie telomerów u jej potomstwa może wpływać też otyłość matki.
Choć istnieją pewne badania sugerujące, że suplementacja koenzymem Q10 może odwrócić skutki skracania telomerów spowodowane nieodpowiednim odżywianiem matki, nie można polegać wyłącznie na suplementacji. Trzeba również dbać o zdrowy styl życia.
Choć genetyka odgrywa pewną rolę w określeniu długości telomerów, to jednak większość (około 80%) wpływu na długość telomerów pochodzi z czynników środowiskowych. Innymi słowy, nasze geny mogą predysponować nas do pewnych cech, ale to nasze zachowania i środowisko w dużej mierze decydują o tym, jak te geny są wyrażane.
Czynniki wpływające na aktywność telomerazy
Telomeraza to enzym odpowiedzialny za dodawanie sekwencji nukleotydów na końce telomerów, co pozwala na ich wydłużenie. Dzięki temu komórki mogą dzielić się przez dłuższy czas, zanim telomery skrócą się do tego stopnia, że komórka przestaje się dzielić lub ulega apoptozie (programowanej śmierci komórki).
Aktywność telomerazy jest kluczowa dla zdrowia komórek i długości życia. W wielu badaniach naukowych wykazano, że czynniki takie jak zdrowa dieta, regularna aktywność fizyczna, unikanie stresu oraz pewne suplementy diety mogą wpływać na aktywność telomerazy, co z kolei może wpływać na długość telomerów i zdrowie komórek.
Warto zwrócić uwagę, że telomeraza nie jest aktywna we wszystkich komórkach ciała. W komórkach somatycznych (większość komórek ciała) telomeraza jest zazwyczaj nieaktywna, co prowadzi do naturalnego skracania telomerów z każdym podziałem komórkowym. Jednak w pewnych komórkach, takich jak komórki macierzyste i niektóre komórki nowotworowe, telomeraza jest aktywna, co pozwala im na nieograniczone podziały.
Dlatego też zrozumienie czynników wpływających na aktywność telomerazy i długość telomerów pozwala zrozumieć procesy starzenia się i chorób związanych z wiekiem. Wspieranie zdrowej aktywności telomerazy może być jednym ze sposobów na promowanie zdrowego starzenia się i opóźnianie procesów związanych z wiekiem.
Dieta w stresie oksydacyjnym
Pewnym wzorcem żywieniowym może być dieta śródziemnomorska czy dieta DASH. Wszyscy wiemy, że możemy znacznie zmienić profil lipidowy pacjenta, ale także możemy spowodować, że będziemy mieli mniejszy marker stresu oksydacyjnego wyrażony w psuciu komórek naszego DNA.
Badania pokazują, że mamy wtedy wyższe poziomy enzymów antyoksydacyjnych i co ważniejsze, spadają nam parametry stresu stanu zapalnego, wyrażone na przykład białkiem ostrej fazy CRP czy profilem cytokin prozapalnych, w tym słynną Interleukina 6 czytelną w TNF-alfa.
Dla odmiany w Western diets profil lipidowy nie wygląda tak, jakbyśmy chcieli. Mamy prawdopodobnie nadmierną masę ciała i za mało dostarczanych antyoksydantów, a co za tym idzie i będziemy sobie gorzej radzić z wolnymi rodnikami, więc będzie większa produkcja cytokin prozapalnych. Co możemy z tym zrobić?
Pomocna może się okazać skala ORAC, która została zaaprobowana przez Narodowy Instytut Zdrowia w Stanach Zjednoczonych, ale coraz częściej implementowana także w innych krajach. W dużym uproszczeniu ta skala mierzy potencjał antyoksydacyjny różnych pokarmów.
Wyraża ona w milimolach na 100 miligram zawartość przeciwutleniaczy w różnych produktach, które są nam bardzo potrzebne do tego, by prawidłowo funkcjonował nasz system antyoksydacyjnym. Jest to na tyle ważny element zdrowia publicznego, że w Stanach Zjednoczonych i Kanadzie etykiety produktów muszą zawierać wskaźnik antyoksydacyjny w skali ORAC. Dzięki temu pacjenci, jedząc produkty, wiedzą ile tych antyoksydantów sobie dostarczają.
Chociaż oznaczenia te nie są stosowane w Polsce, to dotyczy ona produktów naturalnie występujących w każdej szerokości geograficznej. Na liście tzw. „polskich super foods-ów” znajduje się na przykład aronia, jeżyny, czarna porzeczka, śliwki i zioła, a także cynamon z ogromnym potencjałem antyoksydacyjnym.
Drugim pomocnym narzędziem jest skala ANDI, czyli skala gęstości odżywczej, sformułowana przez Joela Furhmana.
Bardzo istotna jest wybieranie do swojej diety tych produktów, które w 100 gramach zawierają dużą gęstość różnych składników, błonnika, zawartości różnych składników B1, B2, B6, B12, C, E, cynku i tak dalej i wielu antyoksydantów. Np. jedząc 100 g frytek, dostarczamy 12, a jak zjemy 100 g jarmużu to 1000.
Suplementacja w stresie oksydacyjnym
Chociaż optymalnym sposobem dostarczania składników odżywczych jest dieta, w pewnych sytuacjach lekarze mogą zalecić suplementację, taką jak fitochemikalia, alkaloidy czy terpenowidy, opartą na konkretnych wynikach badań i dowodach naukowych. W takiej sytuacji ważne jest pamiętanie o kluczowych witaminach i składnikach, takich jak witaminy antyoksydacyjne, koenzym Q 10 i Q 9, oraz o naturalnych produktach, takich jak czosnek czy kurkuma, które mają udowodnione działanie antyoksydacyjne. Wspieranie diety tymi produktami pod kontrolą specjalisty, może pomóc w redukcji stresu oksydacyjnego.
Podsumowanie
W kontekście walki ze stresem oksydacyjnym niezwykle istotne jest dbanie o zdrowie psychiczne i fizyczne. Pomocne mogą się też okazać techniki relaksacyjne, takie jak medytacja czy trening mindfulness, których korzyści w postaci obniżania poziomu cytokin prozapalnych zostały udowodnione.
Ważne jest też prowadzenie aktywnego tryb życia i łączenie wysiłku siłowego z aerobowym, bo przynosi to korzyści dla systemu antyoksydacyjnego. Wszystkie te elementy sprowadzają się do dążenia do równowagi w życiu, dbania o zdrowie psychiczne i fizyczne oraz korzystania z wiedzy i wsparcia specjalistów w dziedzinie zdrowia.
Termin dyspepsja pochodzi z języka greckiego i oznacza „złe trawienie” (dys – złe, peptin – trawienie), często określany jest również jako niestrawność. Z medycznego punktu widzenia jest to zespół objawów klinicznych, które manifestują się dyskomfortem w środkowej części nadbrzusza, uczuciem pełności, czasem bólu. Aby rozpoznać dyspepsję, pacjent musi odczuwać objawy przez co najmniej 4 tygodnie.
Dyspepsja (niestrawność) jest bardzo częstym objawem, ocenia się, że występuje u 15-25% populacji, a wiele osób w ogóle nie zgłasza się z tym problemem do lekarza.
Podstawowe symptomy dyspepsji to:
dyskomfort i/lub ból w nadbrzuszu,
uczucie poposiłkowej sytości,
odbijania, brak łaknienia, zgaga,
nudności, wymioty.
Dyspepsja może być spowodowana przyczynami organicznymi – czyli obecnością procesu chorobowego w przewodzie pokarmowym lub chorobą układową czy metaboliczną. Najczęstszą przyczyną dyspepsji spowodowanej przyczynami organicznymi jest choroba wrzodowa żołądka i dwunastnicy, odpowiada za 15-25% przypadków tego schorzenia. W dalszej kolejności za dyspepsję odpowiada refluks żołądkowo-przełykowy lub dwunastniczo-żołądkowy. Rzadszymi przyczynami są rak żołądka i przełyku, choroby trzustki, hiperkalcemia.
Inną postacią dyspepsji jest dyspepsja czynnościowa, w której objawy nie są spowodowane przyczynami organicznymi. Jest to najczęstsza postać tego schorzenia, 60% przypadków dyspepsji to postać czynnościowa. Uważa się, że za jej występowanie odpowiadają zaburzenia motoryki przewodu pokarmowego, zaburzenia wydzielania kwasu solnego oraz indywidualny próg bólu czucia trzewnego.
Ważną częścią postępowania diagnostycznego jest wywiad lekarski oceniający:
nasilenie objawów (łagodne, umiarkowane, ostre),
czas trwania.
Infekcja Helicobacter pylori
Zakażenie bakterią Helicobacter pylori jest kluczowym czynnikiem ryzyka choroby wrzodowej. Należy jednak podkreślić, że leczenia wymaga nie więcej niż 20% osób zakażonych. W Polsce aż 70% populacji jest nosicielem H.pylori, na świecie ok. 50%.
Infekcja Helicobacter pylori musi być potwierdzona badaniami diagnostycznymi. Zasadą jest, iż u pacjentów powyżej 45 r.ż. oraz u osób z objawami alarmowymi wykonuje się gastroskopię wraz z testem Helicobacter pylori. U pozostałych pacjentów można wykonać antygen w kale lub oddechowy test mocznikowy (UBT). Wykonanie badania wykrywającego antygen w kale pozwala na wykrycie świeżej infekcji. Badanie wykonuje się z próbki kału, jest przydatny nie tylko w diagnostyce infekcji, ale również w monitorowaniu skuteczności leczenia. W tym celu zaleca się jego wykonanie co najmniej po 4 tygodniach od zakończenia eradykacji, w ciągu 8-12 tygodniu.
Oddechowy test mocznikowy, test UBT, to badanie polegające na wykrywaniu aktywności ureazy u pacjentów, którzy uprzednio spożyli znakowany izotopem węgla (nieradioaktywnym C13, lub radioaktywnym C14) mocznik. Jeśli pacjent jest zainfekowany bakterią Helicobacter pylori, która produkuje ureazę, mocznik przetwarzany jest do znakowanego dwutlenku węgla, którego zawartość jest mierzona w wydychanym powietrzu. Należy pamiętać, iż użycie radioaktywnego izotopu węgla nie może mieć miejsca u kobiet ciężarnych oraz u dzieci.
Rekomendacje mówią także o wykonywaniu badań serologicznych, czyli przeciwciał przeciwko Helicobacter pylori w klasie IgG. Przeciwciała pojawiają się po ok. 3-4 tygodniach od zakażenia, jednak utrzymują się długo, nawet po zakończeniu leczenia eradykacyjnego. Z tego powodu badanie ma ograniczoną wartość w rozpoznawaniu świeżej infekcji, a biorąc pod uwagę fakt, iż w Polsce aż 70% populacji może być zakażona, nie powinno być w tym celu stosowane. Ponadto test nie jest rekomendowany do oceny skuteczności leczenia.
Wykrycie świeżej infekcji Helicobacter pylori nie stanowi jeszcze wskazania do jej leczenia, czyli do eradykacji.
Wskazaniami do wyeliminowania Helicobacter pylori są:
wrzody żołądka i dwunastnicy – czynne lub występujące w wywiadzie, w przeszłości,
nowotwory – chłoniak żołądka typu MALT, stan po resekcji żołądka z powodu nowotworu, występowanie raka żołądka w rodzinie,
nasilone zmiany zapalne błony śluzowej żołądka z aftami.
Choroba wrzodowa dwunastnicy i/lub żołądka
Choroba wrzodowa dwunastnicy i/lub żołądka rozpoznawana jest w badaniu gastroskopowym, warunkiem jej wystąpienia jest stwierdzenie nadżerek w endoskopii.
Choroba wrzodowa jest przewlekła, przebiega z okresami zaostrzeń i remisji. Pacjenci najczęściej skarżą się na ból w nadbrzuszu, często jest to ból palący. W przypadku wrzodów żołądka ból pojawia się zwykle po 1-3 godzin po posiłku, natomiast wrzody dwunastnicy charakteryzują się tzw. bólami głodowymi, występującymi na czczo, zwykle wcześnie rano. Bóle ustępują nawet po niewielkim posiłku.
Rozpoznanie wrzodu żołądka w badaniu gastroskopowym wymaga badania kontrolnego po 4-6 tygodniach. Jego celem jest ocena skuteczności leczenia, gojenia się zmian wrzodowych oraz pobranie wycinka do badania histopatologicznego.
Leczenie choroby wrzodowej polega na wyeliminowaniu czynnika sprawczego, czyli bakterii Helicobacter pylori. Istotną częścią postępowania leczniczego jest również terapia niefarmakologiczna, polegająca na unikaniu przez chorych czynników ryzyka takich jak:
Ponadto pacjent powinien spożywać posiłki lekkostrawne, gotowane na parze, przecierane lub zmiksowane. Powinno się unikać potraw smażonych, pieczonych, grillowanych oraz surowych warzyw i owoców.
Leczenie polega na terapii trójlekowej, opartej na amoksycylinie i metronidazolu. Trzecim lekiem jest inhibitor pompy protonowej – IPP. Może to by omeprazol, lanzoprazol, pantoprazol, esomeprazol czy rabeprazol.
Innych schematem leczenia jest terapia poczwórna oparta na tetracyklinie, metronidazolu i bizmucie. W tym schemacie również stosuje się inhibitory pompy protonowej – IPP.
Czasem stosuje się również terapię sekwencyjną, opartą na stosowaniu amoksycyliny w ciągu 1-5 dni, a od 6-12 dnia klarytromycyny z metronidazolem lub tynidazolem. Może być również terapia poczwórna bez bizmutu. Najbardziej optymalny schemat leczenia dobiera lekarz.
Piśmiennictwo
Zasady postępowania w dyspepsji,chorobie wrzodowej i infekcji Helicobacter pylori. Wytyczne Kolegium Lekarzy Rodzinnych w Polsce oraz European Society for Primary Care Gastroenterology (ESPCG). Wytyczne zalecane przez Konsultanta Krajowego w dziedzinie medycyny rodzinnej. Medycyna Praktyczna. Kraków 2016.





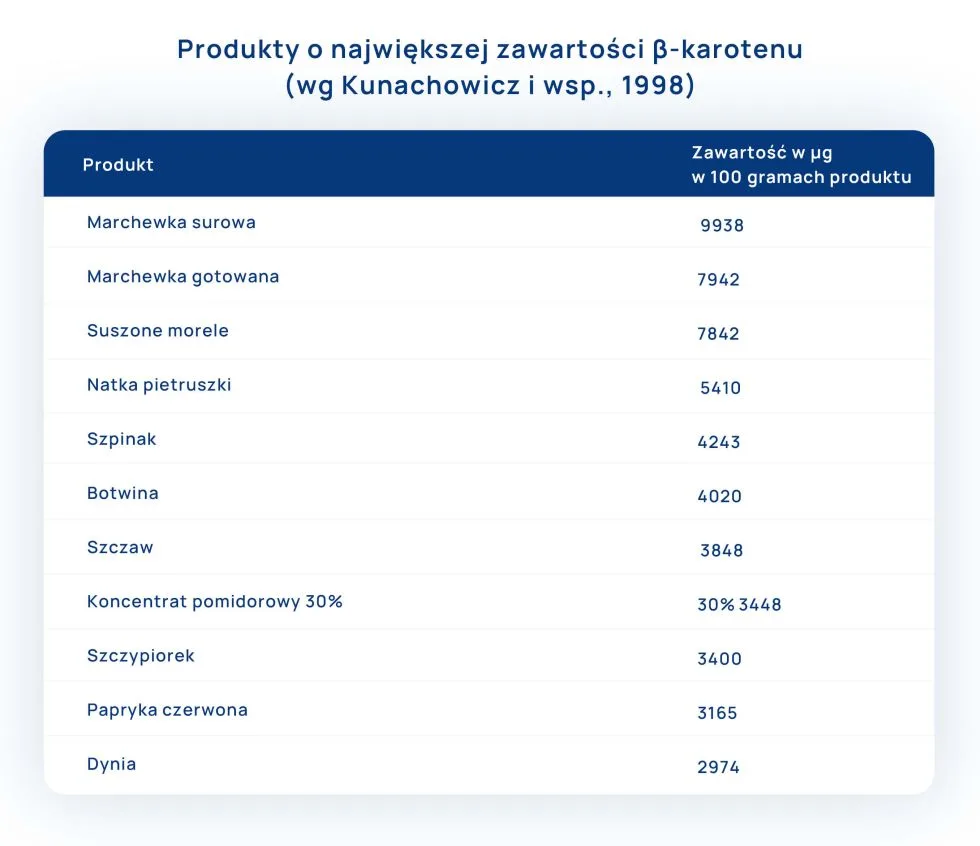
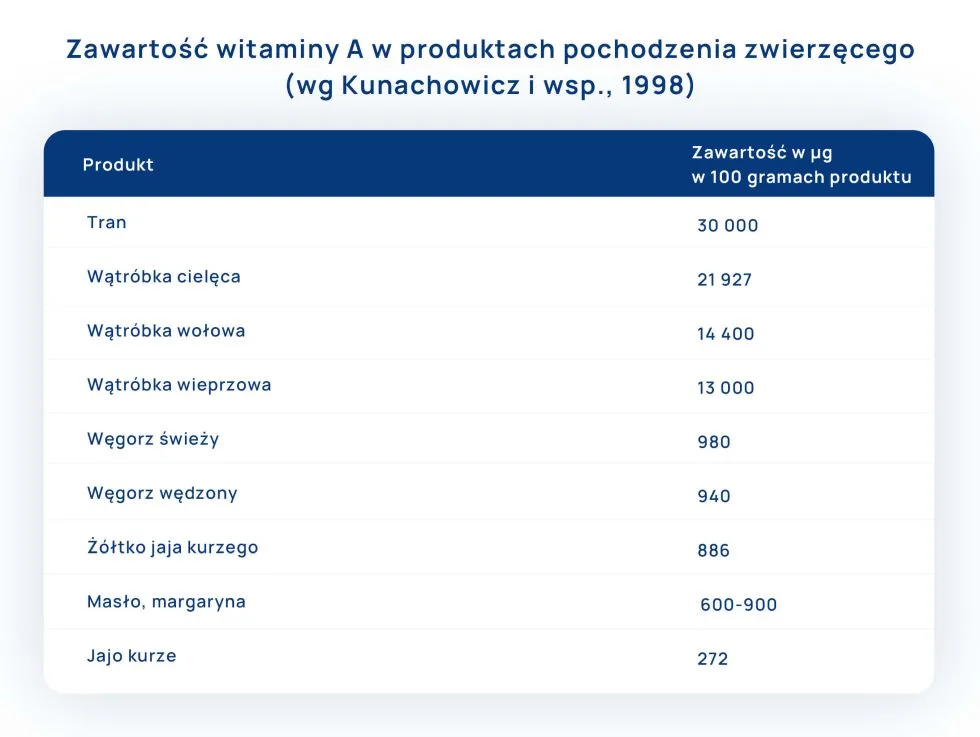
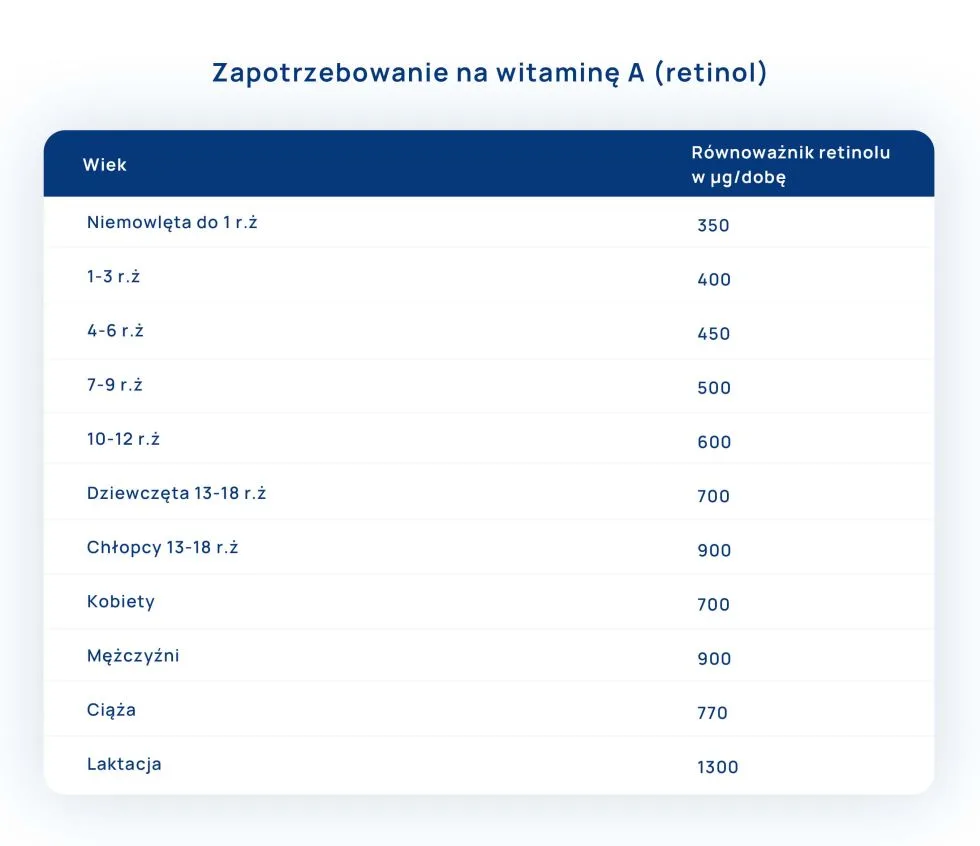
")




")







")