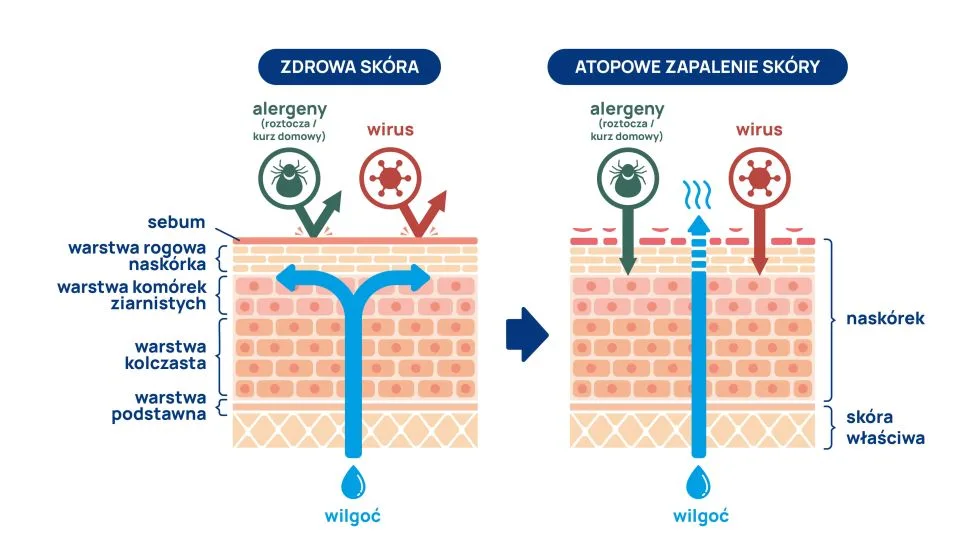
Nerki odgrywają niezwykle istotną rolę w utrzymaniu homeostazy organizmu człowieka. Uczestniczą w metabolizmie wielu hormonów – m.in. kortyzolu, aldosteronu, hormonów płciowych, hormonów tarczycy, katecholamin, prolaktyny, witaminy D3, a także w biodegradacji parathormonu (PTH), kalcytoniny i insuliny.
Zaburzenia endokrynne często pojawiają się w przebiegu przewlekłej choroby nerek (PCHN), ta z kolei staje się coraz powszechniejszym problemem w populacji całego świata. Choroba nerek z uwagi na jej powolny, podstępny, niecharakterystyczny często długo bezobjawowy przebieg stwarza niejednokrotnie wiele problemów diagnostycznych i bywa rozpoznawana zbyt późno. Do najczęstszych przyczyn przewlekłej choroby nerek (PChN) należą: cukrzyca, kłębuszkowe zapalenie nerek, nadciśnienie tętnicze, układowe zapalenia naczyń, wielotorbielowatość nerek dziedziczona autosomalnie dominująco (autosomal dominant polycystic kidney disease, ADPKD), ostre uszkodzenie nerek, do rzadszych zaś sarkoidoza, amyloidoza, zespół Alporta, kolagenozy, zapalenia drobnych naczyń i inne. Choroby te mogą prowadzić do schyłkowej niewydolności nerek (SNN) i konieczności leczenia nerkozastępczego.
Same nerki produkują i wydzielają erytropoetynę, 1,25-OH2-cholekalcyferol i prostaglandyny. Ich niewydolność zaburza te procesy. Zaburzenia endokrynne pogłębiają się wraz z postępem przewlekłej choroby nerek, a w schyłkowej niewydolności osiągają największe nasilenie. Samo nagromadzenie toksyn mocznicowych ma również bezpośredni wpływ na wydzielanie, aktywność i degradację wielu hormonów. Do najistotniejszych zaburzeń na tle endokrynologicznym należą: zespół niskiej trijodotyroniny, subkliniczna i jawna niedoczynność tarczycy, hiperkortyzolemia, hiperprolaktynemia, zwiększone stężenie hormonu wzrostu, hiperinsulinemia, insulinooporność oraz hipogonadyzm.
Hormony przysadki mózgowej
U osób z przewlekłą chorobą nerek stężenie hormonu wzrostu (GH – growth hormone) często oscyluje wokół górnej granicy wartości referencyjnych. Najważniejszą przyczyną jest upośledzona degradacja hormonu wzrostu (GH) przez niewydolne nerki. GH jest ponownie wchłaniany i metabolizowany w kanaliku proksymalnym. Są też liczne doniesienia świadczące o wzmożonej produkcji GH, wskutek niskiego stężenia somatostatyny, albo w odpowiedzi na zwiększone stężenie wydzielanej przez żołądek greliny.
W obrazie klinicznym dominują jednak objawy niedoboru hormonu wzrostu (GH). Dzieje się tak wskutek:
1. Oporności tkanek obwodowych na działania GH
Dokonujące się w większości za pośrednictwem somatomedyn, czyli insulinopodobnych czynników wzrostu – IGF (insulin-like growth factors) 1 i 2 (GH wywiera głównie działanie somatotropowe i metaboliczne poprzez stymulację produkcji insulinopodobnego czynnika wzrostu 1 (insulin-like growth factor 1, IGF-1). W przewlekłej chorobie nerek (PChN) maleje stężenie IGF-1 w surowicy, rosną natomiast stężenia białek wiążących insulinopodobne czynniki wzrostu (IGF-binding proteins, IGFBP) (-1, -2, -4, -6) oraz inhibitora IGF-1). Mimo często prawidłowego stężenia całkowitego IGF ujawniają się liczne zaburzenia ich aktywności, przede wszystkim wskutek obecności osoczowych inhibitorów wiążących IGF →IGF-BP.
2. Wpływu kwasicy metabolicznej oraz współistniejącego stanu zapalnego
Po rozpoczęciu dializoterapii stężenie hormonu wzrostu (GH) spada. Hemodializa prowadzi do wzrostu aktywności IGF-1, prawdopodobnie z uwagi na eliminację niskocząsteczkowych inhibitorów z krwiobiegu. Podawanie rekombinowanej ludzkiej erytropoetyny podczas dializy powoduje również obniżenie stężenia wyjściowego GH.
Po udanym przeszczepieniu nerki czynność osi GH-IGF zwykle się normalizuje.
U dzieci częste są zaburzenia wzrostu wywołane przez nieprawidłowości w układzie GH-IGF (karłowatość nerkowa). U dorosłych obraz kliniczny jest o wiele mniej charakterystyczny. Uważa się, że zaburzenia układu GH-IGF biorą udział – obok przewlekłego procesu zapalnego i niedożywienia – w postępującym wyniszczeniu pacjentów ze schyłkową niewydolnością nerek. Optymalne wydaje wykorzystanie anabolitycznego efektu GH w leczeniu tej grupy pacjentów. Terapia ludzkim GH (human GH, hGH) prowadzi do istotnego wzrostu beztłuszczowej masy ciała. Tendencję wzrostową wykazują osoczowe stężenia albuminy, transferyny oraz cholesterolu – HDL, poprawia się profil lipidowy, następuje spadek stężenia homocysteiny. Ponadto hormon wzrostu (GH) zmniejsza stan zapalny, poprawia stan układu sercowo-naczyniowego oraz zwiększa erytropoezę w różnych populacjach pacjentów. Wykazano, że grelina i GH zwiększają apetyt u niedożywionych i wyniszczonych pacjentów z przewlekłą chorobą nerek.
U dorosłych z przewlekłą chorobą nerek niestety nie oznacza się rutynowo stężenia GH ani IGF-1. Także leczenie, inaczej niż w pediatrii, nie wyszło poza fazę niewielkich badań klinicznych.

Prolaktyna
U ponad połowy pacjentów z zaawansowaną przewlekłą chorobą nerek dochodzi do podwyższenia stężenia prolaktyny. Główną przyczyną podwyższonego stężenia prolaktyny jest zwiększenie jej wytwarzania, choć znaczenie ma też upośledzona biodegradacja. Mechanizmem występującym w przewlekłej chorobie nerek (PCHN) jest istotnie zwiększona sekrecja prolaktyny przez komórki laktotropowe przysadki – zmniejszona dostępność dopaminy w mózgu bezpośrednio stymuluje wydzielanie prolaktyny. Hiperprolaktynemia w przebiegu PChN może dotyczyć nawet połowy chorych w stadium zaawansowanym oraz 30–65% chorych ze schyłkową niewydolnością nerek. Na ogół jest to umiarkowane podwyższenie stężenia prolaktyny, zwykle wynoszące poniżej 100 ng/ml, towarzyszy mu najczęściej zniesienie dobowego rytmu wydzielania hormonu.
W warunkach fizjologicznych prolaktyna wytwarzana jest pulsacyjnie, z nasileniem sekrecji w godzinach wczesnoporannych i najniższymi stężeniami w godzinach wieczornych. W przebiegu przewlekłej choroby nerek (PChN) dochodzi najczęściej do zniesienia dobowego rytmu wydzielania hormonu, spłaszczenia krzywej jego wydzielania oraz braku nocnego spadku stężenia. Wykazano, że stężenie prolaktyny wzrasta wraz z postępem PChN, a najwyższe jego wartości występowały u pacjentów hemodializowanych.
Hiperprolaktynemia w przewlekłej chorobie nerek przebiega najczęściej bezobjawowo, a jej kliniczne manifestacje mogą być mylone z niektórymi objawami choroby podstawowej. Dlatego rozpoznanie kliniczne hiperprolaktynemii w tej grupie pacjentów jest trudne. Jest jednak bardzo ważne, ponieważ prolaktyna jest w stanie modulować odpowiedź zapalną, stymulować adhezję komórek jednojądrzastych do śródbłonka oraz zwiększać proliferację komórek mięśni gładkich naczyń. U niedializowanych pacjentów z przewlekłą chorobą nerek (PChN) wykazano wzrost ryzyka incydentów sercowo-naczyniowych. W takich przypadkach na każde podwyższenie stężenia PRL o 10 ng/ml przypadał wzrost ryzyka incydentów sercowo-naczyniowych nawet o 27%. Hiperprolaktynemia w PCHN nasila wtórne do zmniejszonego stężenia steroidów płciowych objawy hipogonadyzmu (obniżenie libido, zaburzenia erekcji, niepłodność i osteoporozę) oraz zaburzenia miesiączkowania. Zwiększone stężenie prolaktyny jako wynik zmniejszonej aktywności dopaminergicznej oznacza z kolei wzrost wydzielania noradrenaliny, a przez to może mieć niekorzystny wpływ na czynność śródbłonka, sprzyjać nadciśnieniu tętniczemu i w konsekwencji przerostowi mięśnia sercowego. Uważa się również, że hiperprolaktynemia może stanowić mechanizm kompensacyjny przeciwdziałający hipokalcemii (jest jednym z czynników stymulujących syntezę 1,25(OH)2D3).
W diagnostyce hiperprolaktynemii niezbędne jest oznaczenie stężenia prolaktyny. Jeśli znacznie przekracza ono górną granicę wartości referencyjnych, należy szukać innej przyczyny niż przewlekła choroba nerek.
Gonadotropiny
Hipogonadyzm jest częstym powikłaniem przewlekłej choroby nerek (PChN), a zaburzenia funkcji seksualnych ulegają nasileniu w miarę progresji choroby. U pacjentów z PChN hipogonadyzm występuje częściej niż w populacji ogólnej, a jego rozpowszechnienie zawiera się w granicach 40–60%.
U prawie 2/3 mężczyzn leczonych dializami stężenie testosteronu plasuje się poniżej dolnej granicy wartości referencyjnych, przy czym obniżone jest stężenie i całkowitego, i wolnego (aktywnego) testosteronu. Przyczyny obniżonego stężenia testosteronu w przebiegu PCHN nie są do końca wyjaśnione. Są doniesienia dotyczące zwiększonej produkcji oraz nasilonego katabolizmu tego hormonu. U mężczyzn dochodzi do niszczenia jąder z uwagi na toksyczny wpływ mocznicy na spermatogonia. Stężenie wolnego testosteronu może mieć większe znaczenie prognostyczne długoterminowego przeżycia niż wiek. Stężenia całkowitego i wolnego testosteronu są niższe w kwasicy metabolicznej oraz wiążą się ze stężeniami wodorowęglanów. Udowodniono, że obniżone stężenie testosteronu jest czynnikiem ryzyka zgonu z przyczyn sercowo-naczyniowych u pacjentów z PCHN (rozwój miażdżycy), przyczynia się tez do rozwoju niedokrwistości. Niedobór testosteronu prowadzi do zmniejszenia beztłuszczowej masy ciała oraz postępującego wyniszczenia pacjentów w schyłkowej niewydolności nerek. Na typowy obraz zaburzeń składa się podwyższone stężenie lutropiny (LH) i folikulotropiny (FSH), wywołane upośledzoną degradacją, oraz prawidłowe lub nieznacznie obniżone stężenie estradiolu. Nieprawidłowości te obserwowane są już we wczesnych stadiach choroby i nasilają się wraz z jej postępem. Nie ustępują pod wpływem dializoterapii.
Podstawowymi objawami klinicznymi hipogonadyzmu są obniżenie libido, impotencja i niepłodność. Impotencja jest również często skutkiem nie tylko zaburzeń hormonalnych, ale także miażdżycy naczyń obwodowych i niedokrwistości oraz neuropatii, które często towarzyszą przewlekłej chorobie nerek. Zaburzeniom seksualnym w hipogonadyzmie towarzyszyć może również depresja i obniżenie sprawności intelektualnej. Zmniejsza się także masa mięśniowa (beztłuszczowa masa ciała), a zwiększa ilość tkanki tłuszczowej.
W diagnostyce hipogonadyzmu poza obrazem klinicznym istotne są wyniki badań laboratoryjnych, przede wszystkim oznaczenia stężenia testosteronu.
W badaniach stwierdzono, że obniżone stężenie testosteronu przyczynia się do rozwoju niedokrwistości i powikłań sercowo-naczyniowych. Niskie stężenie testosteronu jest także niezależnym czynnikiem ryzyka zgonu z przyczyn sercowo-naczyniowych u pacjentów z przewlekłą chorobą nerek.

Gonadotropiny u kobiet
U kobiet przyczyna zaburzeń cyklu miesiączkowego w przebiegu PChN nie jest do końca jasna. Możliwe, że stanowi ona efekt nieprawidłowej pracy przedniej części przysadki, albo zaburzenia syntezy gonadoliberyny w podwzgórzu. Na neuronach wydzielających gonadoliberynę (GnRH) w podwzgórzu znajdują się receptory dla leptyny, której przypisuje się pewną rolę w zaburzeniach cyklu miesiączkowego w tej grupie chorych. U pacjentek ze skrajną niewydolnością nerek dochodzi również do obniżenia klirensu opioidów i podwyższenia stężenia krążących endorfin, czego rezultatami mogą być zmniejszenie wydzielania GnRH i hamowanie owulacji. Zaawansowana przewlekła choroba nerek prowadzi do zaburzeń miesiączkowania u większości kobiet. W badaniach laboratoryjnych stwierdza się najczęściej podwyższone stężenie LH, nie dochodzi jednak z reguły do przedowulacyjnego wzrostu stężenia tego hormonu. Stężenie folikulotropiny (FSH) pozostaje w granicach wartości prawidłowych. Stężenie estradiolu jest niskie, często poniżej dolnej granicy wartości referencyjnych.
Do typowego obrazu klinicznego należą zaburzenia miesiączkowania, cykle bezowulacyjne, bezpłodność i obniżenie libido. Zaburzenia miesiączkowania to jedne z pierwszych objawów zaburzeń hormonalnych u pacjentek z PChN. Są częste (73%) u kobiet przed menopauzą ze skrajną niewydolnością nerek, przy czym ponad połowę przypadków stanowi brak miesiączki. W miarę postępu PChN miesiączka staje się bardziej skąpa, a cykle menstruacyjne – nieregularne. Zwykle do braku miesiączki dochodzi jeszcze przed rozpoczęciem przez chorą dializoterapii. U kobiet, które do momentu rozpoczęcia dializ miesiączkują regularnie, zaburzenia te pojawiają się nieco później. Z kolei metro- oraz menorrhagia często prowadzą do znacznej utraty krwi, a niekiedy nawet do konieczności przeprowadzenia transfuzji.
Diagnostyka zaburzeń gonadalnych u kobiet z przewlekłą chorobą nerek opiera się na laboratoryjnej ocenie stężeń hormonów: LH, FSH, estradiolu. Do powikłań, poza zaburzeniami seksualnymi, należy zmniejszenie gęstości mineralnej kości, prowadzące do osteoporozy.

Hormony nadnerczy
Glikokortykosteroidy
Czynność osi podwzgórze-przysadka-nadnercza w przewlekłej chorobie nerek (PCHN) jest zaburzona jedynie częściowo. Stężenia w surowicy oraz rytm dobowy wydzielania kortyzolu i hormonu adrenokortykotropowego (adrenocorticotropic hormone, ACTH) są z reguły niezmienione lub nieznacznie zwiększone. Zwiększone stężenie kortyzolu u chorych z PChN występuje częściej w przypadku jednoczesnego rozpoznania nadciśnienia tętniczego samoistnego. Nerki odgrywają istotną rolę w metabolizmie glikokortykosteroidów, ponieważ biorą udział w usuwaniu z organizmu kortyzolu i jego metabolitów rozpuszczalnych w wodzie oraz przy pomocy enzymu dehydrogenazy 11β-hydroksysteroidowej typu 2 przekształcają biologicznie aktywny kortyzol do jego nieaktywnej formy – kortyzonu. Zapobiega to nadmiernej aktywacji receptora mineralokortykosteroidowego przez kortyzol, co zapewnia ochronę przed nadmierną retencją sodu i związanym z tym rozwojem nadciśnienia tętniczego. Aktywność dehydrogenazy w PChN jest zmniejszona, co skutkuje wydłużonym czasem półtrwania kortyzolu we krwi oraz akumulacją jego metabolitów. Dodatkowo stężenie wolnego kortyzolu jest większe niż stężenie kortyzolu całkowitego w wyniku zmniejszonego wiązania kortyzolu z albuminami, przy jednoczesnym niezmienionym wiązaniu z transkortyną (globuliną wiążącą kortyzol).
U chorych dializowanych, w porównaniu z pacjentami leczonymi zachowawczo, obserwowano zmniejszone nasilenie hiperkortyzolemii, obniżone stężenie ACTH oraz normalizację czasu półtrwania kortyzolu w surowicy. Rytm dobowy wydzielania kortyzolu jest z reguły niezmieniony. Hiperkortyzolemia w przebiegu PChN nie wymaga leczenia przyczynowego, a jedynie uważnego kontrolowania jej powikłań, takich jak: nadciśnienie tętnicze, cukrzyca, osteopenia czy otyłość brzuszna. Zwiększone stężenie kortyzolu w surowicy koreluje z częstością występowania kacheksji i może być jedną z przyczyn częstszych hospitalizacji. W przewlekłej chorobie nerek nieprawidłowy jest test z deksametazonem z powodu upośledzonego wchłaniania deksametazonu z przewodu pokarmowego (sekrecja ACTH nie może być hamowana przez stosowanie standardowych doustnych dawek deksametazonu). Należy o tym pamiętać w diagnostyce zespołu i choroby Cushinga.

Mineralokortykoidy
Stężenie aldosteronu u pacjentów z przewlekłą chorobą nerek jest zwykle prawidłowe lub obniżone, co wynika z niskiej aktywności reninowej osocza. Upośledzony jest też wzrost wydzielania aldosteronu w odpowiedzi na hipowolemię. Do wzrostu wydzielania i podwyższenia stężenia aldosteronu dochodzi tylko w przebiegu zespołu nerczycowego. U pacjentów dializowanych czynnikiem hamującym wydzielanie aldosteronu jest heparyna.
W przewlekłej chorobie nerek aldosteron bierze udział w regulacji stężenia potasu w stopniu znacznie większym niż w populacji ogólnej, stosowanie leków interferujących z syntezą i aktywnością aldosteronu może się więc wiązać z groźną dla życia hiperkaliemią i dlatego powinno być prowadzone z dużą ostrożnością.
Aminy katecholowe
Stężenie amin katecholowych w przewlekłej chorobie nerek jest zwykle wyższe niż w populacji ogólnej, na co wpływa spadek klirensu nerkowego i zmniejszenie degradacji. Uważa się, że katecholaminy mogą mieć udział w patogenezie nadciśnienia tętniczego w chorobach nerek.
Hormony tarczycy
W przewlekłej chorobie nerek (PChN) dochodzi do zaburzeń metabolizmu obwodowego, degradacji i wydalania hormonów tarczycy, co powoduje zaburzenia osi podwzgórze–przysadka–tarczyca i istotnie wpływa na czynność tego narządu.
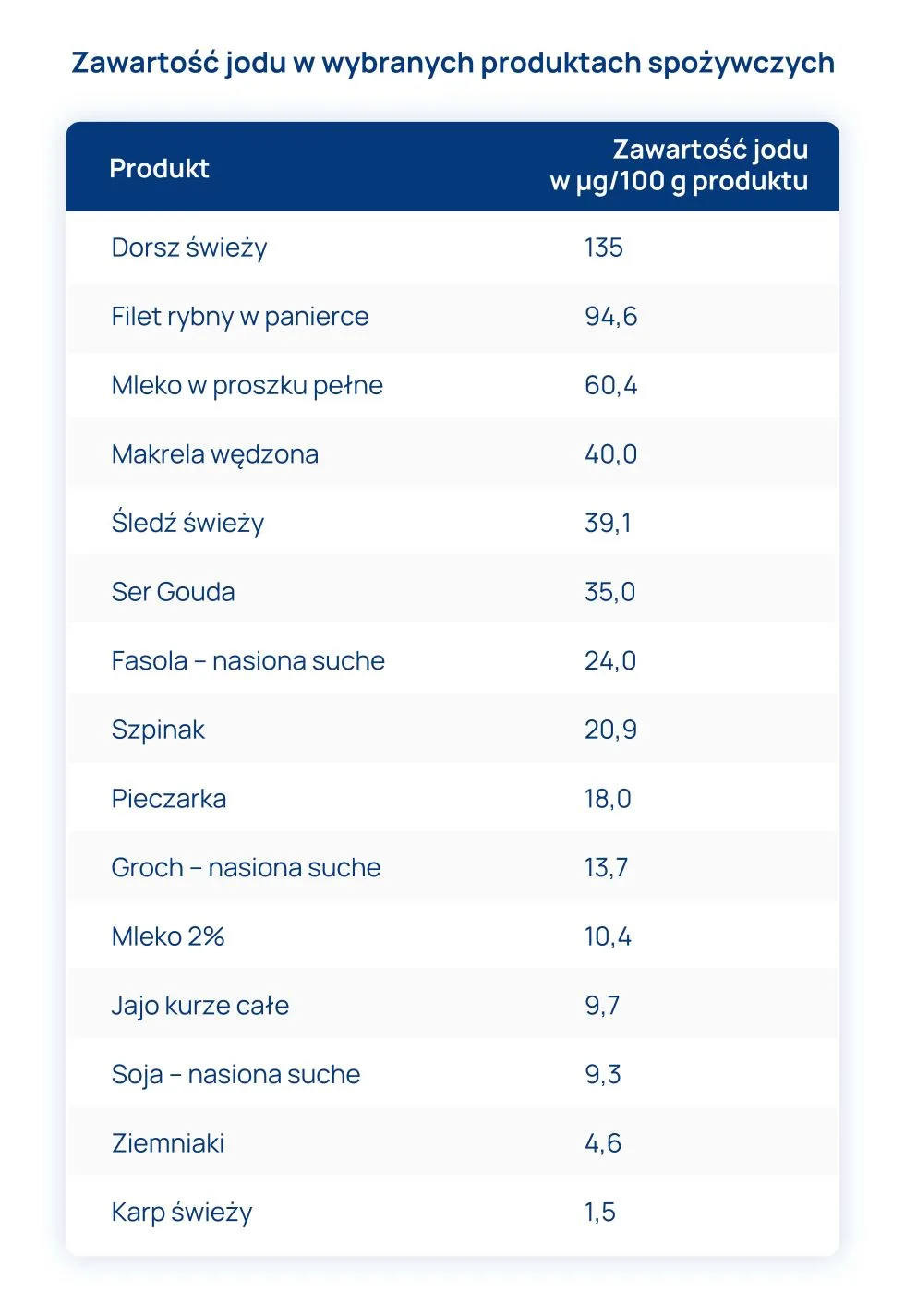
W przebiegu PChN pojawia się narastająca kwasica metaboliczna, wyniszczenie oraz niedożywienie białkowo-energetyczne, które prowadzą do nieprawidłowości dobowego rytmu wydzielania tyreotropiny – TSH (hormon przysadki), co skutkuje również zmniejszonym stężeniem trijodotyroniny – T3, zaburzeniami konwersji tyroksyny – T4 do T3, zmienionym metabolizmem nerkowym, zmianami stężeń białek wiążących hormony, spadkiem zawartości hormonów w tkance tarczycowej oraz zwiększonym gromadzeniem jodu w tarczycy. Stężenia jodu w surowicy wzrasta, przede wszystkim wskutek upośledzonego jego wydalania, a wysoki poziom jodu również hamuje wydzielanie hormonów tarczycy.
Stężenie T4 pozostaje w granicach wartości referencyjnych lub jest nieznacznie obniżone (oznaczane są wolne frakcje hormonów tarczycy – fT4, fT3, niezwiązane z białkowymi nośnikami, ponieważ jedynie forma niezwiązana jest aktywna metabolicznie). Do najczęstszych zaburzeń endokrynologicznych w przewlekłej chorobie nerek (PChN) należy zespół niskiej T3, który u pacjentów z zaburzeniem funkcji nerek charakteryzuje się niskim stężeniem T3 z prawidłowymi stężeniami TSH oraz odwrotnej trijodotyroniny – rT3, co istotnie różnicuje pacjentów bez współistniejącej choroby nerek.
Zarówno subkliniczna, jak i jawna niedoczynność tarczycy występuje u 20% chorych ze wskaźnikiem filtracji kłębuszkowej (GFR) poniżej 60 ml/min/1,73 m2 , a odsetek ten wzrasta wraz z progresją choroby nerek. W stosunku do populacji ogólnej w tej grupie chorych częściej (nawet do 35%) obserwuje się także zwiększenie objętości tarczycy, występowanie rozlanego wola (szczególnie u kobiet) oraz raka tarczycy.
U pacjentów z niskim stężeniem trijodotyroniny (T3) i wolnej trijodotyroniny (fT3) nie obserwuje się z reguły objawów hipotyreozy, być może z powodu zwiększenia ekspresji receptorów dla fT3. Nadczynność tarczycy występuje natomiast z taką samą częstością jak w populacji ogólnej.
Choroby autoimmunizacyjne tarczycy również występują z taką samą częstością u chorych z PChN i w populacji ogólnej. Objawy kliniczne hipotyreozy są podobne do objawów przewlekłej choroby nerek, dlatego trudno ją rozpoznać na podstawie obrazu klinicznego. W diagnostyce zaburzeń endokrynnych związanych z gruczołem tarczowym konieczne jest oznaczenie hormonów tarczycowych – ich wolnych frakcji fT3 i fT4 oraz TSH- hormonu przysadki. Leczenie nie różni się istotnie od stosowanego u osób z prawidłową funkcją nerek
Obniżenie stężenia T3 i tym samym fT3 jest niezależnym czynnikiem ryzyka zgonu. U kobiet z przewlekłą chorobą nerek większe jest ryzyko wystąpienia guzków tarczycy i raka tarczycy.
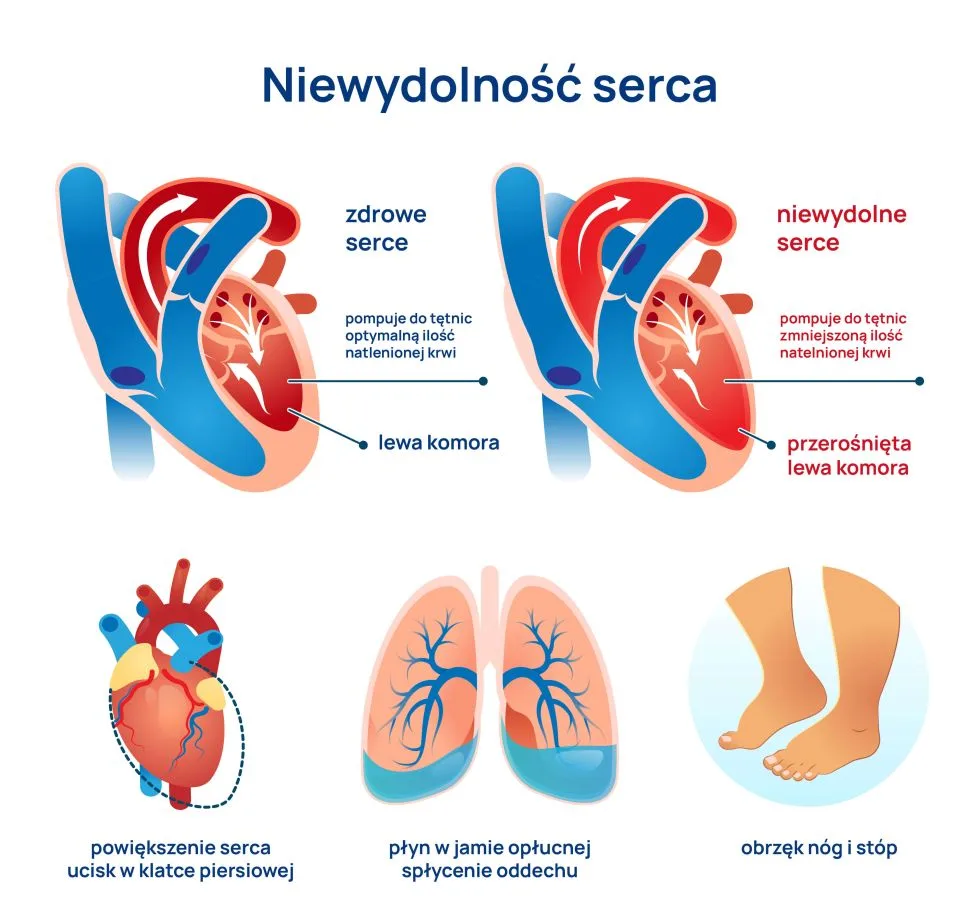
Zaburzenia endokrynne występują często u chorych z przewlekłą chorobą nerek, pogłębiają się wraz z postępem choroby, a ich największe nasilenie obserwuje się w fazie schyłkowej niewydolności nerek. Stanowią również ważny problem kliniczny, ponieważ istotnie wpływają na jakość życia, zwiększają też liczbę powikłań i zgonów. Wymagają zatem szybkiej diagnostyki i odpowiedniego leczenia. Leczenie nerkozastępcze (dializy) nie ma istotnego wpływu na większość zaburzeń endokrynnych. Normalizacji tych zaburzeń można spodziewać się dopiero po udanej transplantacji nerki.
Piśmiennictwo
- Wielka Interna, Nefrologia: M. Chmielewski, B. Rutkowski: Zaburzenia endokrynne w przewlekłej chorobie nerek.
- J. Sobolewska, Z. Żak, K. Monia-Tutur, A. Wojciechowska-Luźniak, P.Witek, S Niemczyk: Zaburzenia endokrynologiczne w przewlekłej chorobie nerek, Pediatr.Med.Rodz. 2022,18(3)












 u dzieci")


")


")