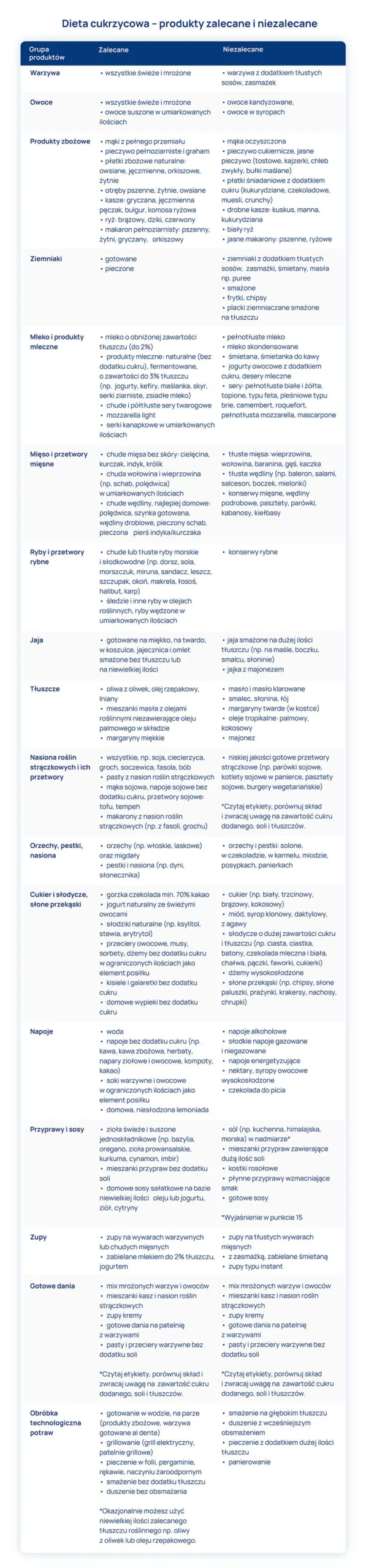
")
Spis treści:
- Czym jest zespół jelita drażliwego?
- Dieta w chorobie zespołu jelita drażliwego
- Zalecenia dietetyczne w zespole jelita drażliwego
- Przykładowy jadłospis w zespole jelita drażliwego
Czym jest zespół jelita drażliwego?
Zespół jelita drażliwego, inaczej zwany IBS (irritable bowel syndrome), to zaburzenie czynnościowe jelita cienkiego i jelita grubego. Objawia się ono dyskomfortem lub bólem brzucha oraz zaburzeniami rytmu wypróżnień. Przy czym objawy te nie są uwarunkowane zmianami organicznymi czy biochemicznymi.
>> Jak zdiagnozować zespół jelita drażliwego, jakie badania wykonać? Sprawdź w artykule: Zespół jelita drażliwego – objawy, diagnostyka, leczenie
Dieta w chorobie zespołu jelita drażliwego
W zespole jelita drażliwego nie ma jednolitych zaleceń dietetycznych dla wszystkich chorych. Przebieg IBS jest przewlekły. W zależności od dominującego objawu może występować postać biegunkowa, postać mieszana lub zaparciowa.
Leczenie obejmuje odpowiednie postępowanie dietetyczne, dostosowane do potrzeb pacjenta oraz leczenie wspomagające farmakologiczne. Niestety, u większości chorych objawy nawracają, co znaczy, że rokowania co do całkowitego wyleczenia są niepomyślne.
Na początku należy ocenić dotychczasowy sposób żywienia chorego z zespołem jelita drażliwego. Trzeba ustalić:
- regularność spożywanych posiłków,
- ilość spożywanych posiłków,
- rodzaj najczęściej spożywanych produktów spożywczych, potraw i napojów
- sposoby przygotowania potraw, rodzaje technik (gotowanie, pieczenie, smażenie, grillowanie itd.),
- spożywanie kawy,
- spożywanie napojów alkoholowych,
- aktywność fizyczną.
Kolejnym krokiem jest przygotowanie przez pacjenta kalendarzyka żywieniowego, który powinien zawierać:
- porę pobudki,
- godziny spożycia poszczególnych posiłków – dokładne wyszczególnienie rodzaju i wielkości spożytych pokarmów i wypitych napojów, jaki był czas posiłku i w jakiej atmosferze był spożywany,
- notatki na temat samopoczucia pacjenta po spożyciu każdego posiłku – czy występowały dolegliwości, a jeśli tak, to jakie.
Analiza informacji będzie pomocna w identyfikacji produktów pokarmowych wywołujących i nasilających objawy u chorego.

Następnym etapem jest eliminacja nieprawidłowych nawyków żywieniowych oraz wprowadzenie do diety pacjenta zindywidualizowanych zaleceń żywieniowych. Uwzględniamy objawy chorobowe oraz zwyczaje żywieniowe.
W pierwszym etapie eliminujemy z diety pacjenta produkty, które nasilały dolegliwości ze strony zespołu jelita drażliwego. W przypadku, gdy takie postępowanie nie przynosi efektu, stosujemy dietę lekkostrawną:
- chude mięso,
- ryż,
- niektóre owoce i warzywa.
Następnie powoli rozszerzamy dietę o kolejne produkty, w odstępach czasowych, co kilka dni.
Chory notuje każde kolejno wprowadzane produkty oraz zapisuje, czy wystąpiły w tym czasie jakieś dolegliwości. Postępowanie to prowadzi do identyfikacji produktów spożywczych, które przyczyniają się do zaostrzenia objawów IBS. Wykluczenie ich z diety pozwala na zmniejszenie nasilenia dolegliwości u części pacjentów.
Ze względu na duży dyskomfort codziennego życia z zespołem jelita nadwrażliwego, znaczna część pacjentów eliminuje z diety niektóre produkty, potrawy i napoje zupełnie niezależnie od rzeczywistego związku ich spożycia z nasileniem dolegliwości.
Aby zachować różnorodność diety, należy cierpliwie analizować sposób żywienia chorych z IBS i stopniowo wprowadzać kolejne produkty. To pozwoli nam na wyeliminowanie tych produktów, które najbardziej nasilają objawy choroby u danego pacjenta.
Wprowadzając prawidłową dietę, należy pamiętać o:
- odpowiedniej objętości posiłków,
- jaka jest gęstość energetyczna posiłku w krótkim czasie,
- zapobieganiu kompulsywnemu jedzeniu, które prowadzi do zwiększenia objętości treści pokarmowej w jelitach,
- dostarczeniu dużej ilości makroskładników do organizmu, które w krótkim czasie prowadzi do:
- zaburzeń trawienia i wchłaniania,
- nadmiernej fermentacji resztek pokarmowych w okrężnicy,
- występowania wzdęć oraz bólu,
- ocenie, czy w diecie występują produkty sprzyjające powstawaniu wzdęć.
>> Sprawdź też: Ból brzucha – przyczyny, lokalizacje, diagnostyka (alablaboratoria.pl)
Czego nie jeść w zespole jelita drażliwego?
W zespole jelita wrażliwego powinniśmy unikać:
1) produktów wzdymających – cebula, czosnek, kapusta, kalafior, brokuł, brukselka, kukurydza, rośliny strączkowe, papryka, orzechy, suszone śliwki, słodycze, napoje gazowane;
2) innymi produktami dającymi częste objawy w zespole jelita drażliwego są – produkty mleczne, produkty pszenne, jaja, owoce cytrusowe, drożdże, ostre przyprawy, produkty wysoko przetworzone jak pizza, typu fast food;
3) substancji słodzących, które są stosowane w napojach, gumach do żucia oraz słodyczach. Polialkohole – sorbitol, ksylitol i mannitol oraz fruktoza – spożywane w dużych ilościach, mogą spowodować biegunki. Należy ograniczyć spożywanie produktów je zawierających;
4) kawy i alkoholu, które w zespole jelita drażliwego są często źle tolerowane. Należy ograniczyć picie kawy i produktów alkoholowych w chorobie IBS, a w przypadku zwiększonych dolegliwości całkowicie je wyeliminować z diety;
5) błonnika pokarmowego – w dużych ilościach błonnik pokarmowy może prowadzić do biegunek, dlatego należy obserwować jego podaż wraz z dietą u chorych ze skłonnością do występowania biegunki.
Mleko i jego przetwory jest często eliminowane z diety przez chorych. Wynika to z kojarzenia nietolerancji laktozy z dolegliwościami w IBS. Często takie postępowanie prowadzi do niewystarczającej podaży wapnia w diecie i zmniejsza urozmaicenie spożywanych produktów.
Badania nie wykazały, aby nietolerancja laktozy występowała częściej u chorych na zespół jelita nadwrażliwego niż u osób niedotkniętych tym schorzeniem. Trzeba nadmienić, że objawy w nietolerancji laktozy i IBS są podobne. Nie należy eliminować mleka i produktów mlecznych u osób z IBS, należy się obserwować. Można zastosować żywność o zmniejszonej zawartości laktozy, czyli fermentowane produkty mleczne.
Produkty wskazane w diecie IBS (irritable bowel syndrome)
Jeżeli pomimo przestrzegania ogólnych zasad stylu życia i modyfikacji żywienia według indywidualnej tolerancji objawy IBS utrzymują się, należy rozważyć wprowadzenie diety eliminacyjnej.
Dalsze postępowanie dietetyczne często obejmuje zastosowanie diety o niskiej zawartości FODMAP czyli z wyeliminowaniem fermentujących oligosacharydów, disacharydów, monosacharydów i polioli.
Związki te mogą nasilać objawy zespołu jelita nadwrażliwego poprzez zwiększenie ilości płynu w jelicie cienkim, nadmierną fermentację, przyspieszenie pasażu jelitowego i zwiększenie wydzielania gazów.
Dieta low-FODMAP powinna być prowadzona pod opieką specjalisty, przez okres nie dłuższy niż około 4-6 tygodni, ponieważ zakłada duże restrykcje dietetyczne niekorzystnie wpływa na skład mikroflory jelit.
Dieta low-FODMAP zakłada 3 etapy:
- Etap 1 całkowitej eliminacji produktów o wysokiej zawartości FODMAP,
- Etap 2 stopniowego rozszerzania diety o produkty FODMAP.
- Etap 3 wprowadzany na końcu, zakładający wykluczenie jedynie tych produktów FODMAP, po których w organizmie pacjenta wciąż utrzymywały się dolegliwości z etapu 2.
Dieta low-FODMAP może pomóc zmniejszyć lub wyeliminować objawy od strony układu pokarmowego. Może przyczynić się do poprawy jakości życia poprzez zmniejszenie uczucia wzdęcia i dyskomfortu, poprawę rytmu wypróżnień.
Nie dzieje się tak jednak u każdego chorego. Pomaga ona około 70% pacjentów z IBS. Wpływ na obniżenie skuteczności tej diety ma między innymi nieuważne jej przestrzeganie w 1 etapie, dlatego powinna być prowadzona pod okiem specjalisty.
U pacjentów z IBS nie rekomenduje się natomiast wprowadzania innych nieuzasadnionych klinicznie diet eliminacyjnych, np. bezglutenowej czy bezlaktozowej. Każda restrykcyjna dieta eliminacyjna może skutkować wystąpieniem niedoborów pokarmowych i niedożywieniem, co ma wpływ na pogorszenie samopoczucia i ogólnego stanu zdrowia.
Probiotykoterapia jest bardzo ważnym elementem powrotu do prawidłowej pracy jelit w zespole jelita nadwrażliwego. Polega na włączeniu suplementacji probiotykami, czyli wybranymi szczepami żywych drobnoustrojów podawanymi w odpowiednich ilościach. Wpływają one korzystnie na skład mikrobioty jelit, bo zwiększają udział „dobrych” bakterii jelitowych.
Pacjenci z zespołem jelita nadwrażliwego często narażeni są na dysbiozę jelit, która może być jedną z przyczyn powstawania tej choroby oraz nasilenia jej objawów. Według stanowiska World Gastroenterology Organisation sugeruje się, że niektóre probiotyki (np. szczep Lactobacillus plantarum 299v) podane w odpowiedniej dawce mogą łagodzić objawy i poprawiać jakość życia osób z IBS. Rodzaj probiotyków oraz czas suplementacji powinien być ustalany indywidualnie według wskazań lekarza. Nie należy samodzielnie wdrażać suplementacji.

Zalecenia dietetyczne w zespole jelita drażliwego
Zalecenia dietetyczne w IBS:
- regularne spożywanie pięciu posiłków, w stałych godzinach, niezbyt obfitych,
- wykluczenie z diety produktów źle tolerowanych – obserwacja indywidualnej oceny tolerancji,
- ograniczenie w diecie tłustych produktów spożywczych,
- eliminacja z diety produktów i potraw wzdymających,
- eliminujemy z diety substancje słodzące polialkohole i duże ilości fruktozy,
- ocena udziału w diecie dostarczanego błonnika pokarmowego oraz jego źródła, modyfikacja jego ilości w zależności od występujących objawów chorobowych (biegunki, zaparcia),
- ograniczenie lub całkowite wyeliminowanie kawy, mocnej herbaty, napoi alkoholowych,
- eliminacja ostrych przypraw,
- u osób z nietolerancją laktozy, ograniczamy jej ilość w diecie do poziomu dobrze tolerowanego przez pacjenta,
- eliminacja z diety napojów gazowanych,
- wprowadzenie aktywności fizycznej, dostosowanej do możliwości pacjenta,
- do picia woda niegazowana, spożywana regularnie,
- przygotowanie potraw to gotowanie w wodzie, przyrządzanie na parze, pieczenie w folii, duszenie bez tłuszczu lub z małym dodatkiem tłuszczu,
- unikamy smażenia/duszenia w głębokim tłuszczu, w panierkach, grillowania i pieczenia w sposób tradycyjny.
Ważne, aby wprowadzanie do diety chorego nowego produktu było skrupulatnie odnotowywane. Pomoże to ustalić stopień tolerancji danego produktu spożywczego przez organizm chorego. Produkty żywieniowe wprowadzamy pojedynczo, w kilkudniowych odstępach czasu.
>> Przeczytaj: Nietolerancja węglowodanów – przyczyny, objawy, diagnostyka (alablaboratoria.pl)
Przykładowy jadłospis w zespole jelita drażliwego
I śniadanie
Płatki owsiane z mlekiem migdałowym, bananem i truskawkami.
II śniadanie
Kromka pieczywa bezglutenowego z wędliną z indyka do tego mix sałat i ogórek zielony.
Obiad
Udziki z kurczaka bez skóry, obtoczone w delikatnych ziołach, pieczone w piekarniku w rękawie.
Do tego komosa ryżowa oraz gotowana marchewka.
Podwieczorek
Marchew i ogórek surowe, obrane i pokrojone w słupki. Do tego jogurt naturalny bezlaktozowy z dodatkiem ulubionych delikatnych ziół. Warzywa maczamy w dipie.
Kolacja
Pieczywo z mąki orkiszowej posmarowane masłem. Mozzarella pokrojona w plastry, ułożona na pomidorach pokrojonych w plastry, posypane delikatnie bazylią.
Źródła:
- Adrych K., Rydzewska G.: Rozpoznawanie i leczenie zespołu jelita nadwrażliwego w praktyce lekarza rodzinnego. Varia Medica 2020;4(1):52-59.
- Włodarek D.: Dietoterapia, 8.Wybrane choroby przewodu pokarmowego. Wydanie I – 3 dodruk, Warszawa 2015, Wydawnictwo Lekarskie PZWL
- Moayyedi P., Andrews CN., MacQueen G. i wsp.: Canadian Association of Gastroenterology Clinical Practice Guideline for the Management of Irritable Bowel Syndrome (IBS). Journal of the Canadian Association of Gastroenterology, 2019; 2(1): 6–29. doi: 10.1093/jcag/gwy071
- Monash University: FODMAP and IBS [online][dostęp: 15.02.2023]. Dostępny w https://www.monashfodmap.com/about-fodmap-and-ibs/
- Vasant DH., Paine PA., Black CJ. i wsp.: British Society of Gastroenterology guidelines on the management of irritable bowel syndrom. Gut 2021;70:1214–1240. doi:10.1136/gutjnl-2021-324598






 – jak objawia się kandydoza?")











: przyczyny, objawy i leczenie")

: podwyższona, za niska – co to oznacza i jak interpretować wyniki?")
