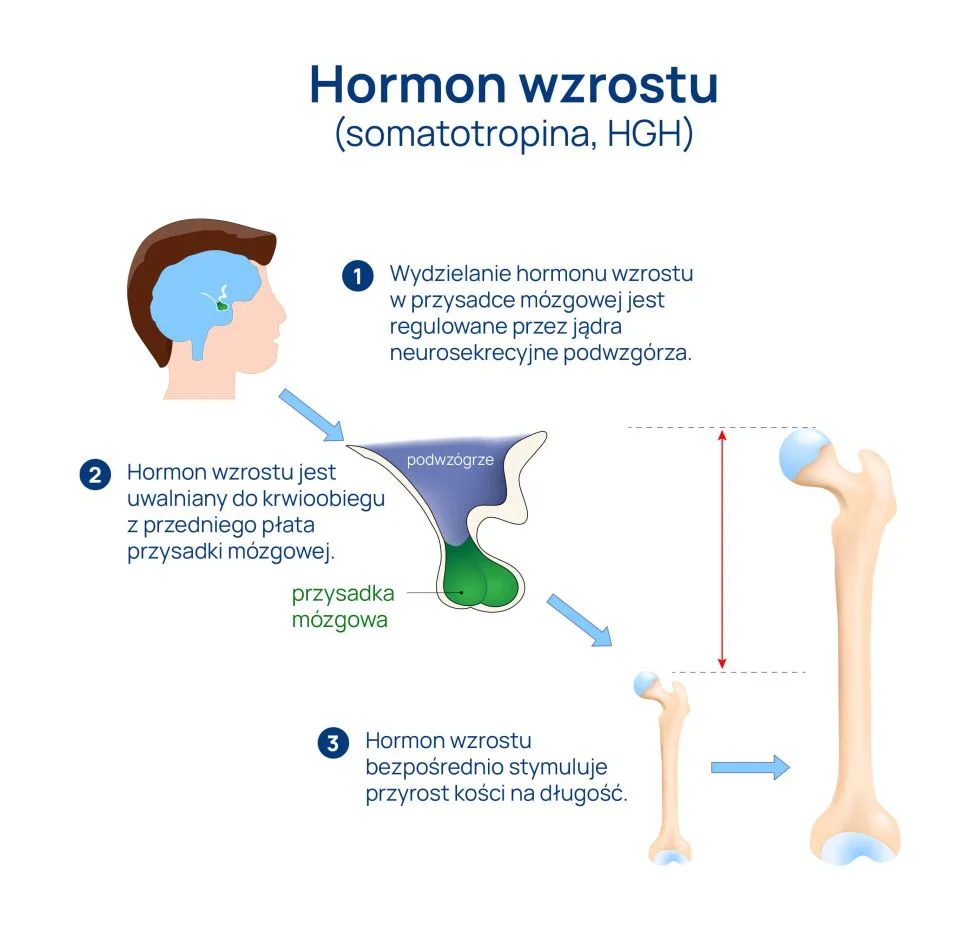
Testy na ojcostwo są szeroko stosowane w celu potwierdzenia biologicznego pokrewieństwa między ojcem a potomstwem. Wykorzystują one zaawansowane techniki analizy genetycznej, aby precyzyjnie określić, czy osoba jest biologicznym rodzicem dziecka. Takie testy są istotne nie tylko w kontekście rozstrzygania spraw prawnych, ale także w diagnostyce genetycznej. W poniższym artykule omówimy zasadę działania testów na ojcostwo, a także wskazówki dotyczące optymalnego momentu na ich przeprowadzenie.
Spis treści:
- Na czym polega test na ojcostwo?
- Kiedy można zrobić test na ojcostwo?
- Czy testy na ojcostwo są wiarygodne?
Na czym polega test na ojcostwo?
Test na ojcostwo, znany również jako test DNA na pokrewieństwo, polega na analizie materiału genetycznego pobranego od potencjalnego ojca i dziecka. Badanie to ma na celu porównanie profili genetycznych obydwu osób, aby określić, czy istnieje biologiczne pokrewieństwo między nimi. Testy te opierają się na analizie markerów genetycznych, które są przekazywane z pokolenia na pokolenie. W takich badaniach stosuje się różne techniki, w tym analizę polimorfizmów długości fragmentów restrykcyjnych (RFLP) czy zmienną liczbę powtórzeń tandemowych (VNTR) oraz inne modyfikacje techniki PCR, aby uzyskać wyniki o wysokiej precyzji.
| Czy wiesz że? Testy na ojcostwo nie są ograniczone tylko do ludzi. W hodowli zwierząt, zwłaszcza w przypadku rasowych psów i kotów, testy genetyczne są stosowane do potwierdzania linii rodowodowych oraz jakości hodowli. Wykorzystują one podobne technologie do tych stosowanych u ludzi. |
Jak wygląda test na ojcostwo?
Test na ojcostwo jest stosunkowo prosty do wykonania. Najczęściej polega na pobraniu próbki materiału biologicznego, takiego jak krew, ślina lub komórki nabłonkowe z jamy ustnej, od ojca i dziecka. W przypadku testów nieinwazyjnych, które są coraz bardziej popularne, można również wykorzystać próbki krwi matki do izolacji wolnego płodowego DNA. Po pobraniu próbek oraz wyizolowaniu z nich DNA, laboratoria genetyczne przeprowadzają analizę, porównując sekwencje genetyczne i szukając zgodności między markerami dziedziczonymi od ojca i dziecka.
Gdzie zrobić test na ojcostwo?
Testy na ojcostwo można wykonać w wielu miejscach, w tym w specjalistycznych laboratoriach genetycznych, ośrodkach diagnostycznych, klinikach medycznych oraz za pośrednictwem firm zajmujących się badaniami DNA. Ważne jest, aby wybierać renomowane laboratoria, które stosują nowoczesne technologie i zapewniają wiarygodne wyniki.



Ile kosztuje test na ojcostwo?
Koszt testu na ojcostwo może się znacznie różnić w zależności od wybranej metody oraz laboratorium. Standardowe testy wykonywane po narodzinach dziecka kosztują zazwyczaj od kilkuset złotych do kilku tysięcy złotych. W przypadku testów nieinwazyjnych przeprowadzanych przed narodzinami potomstwa,, koszt może wynosić nawet kilka tysięcy złotych. W kontekście spraw sądowych, gdzie test na ojcostwo wymaga dodatkowych formalności, takich jak potwierdzenie wyników w sądzie, ceny mogą być jeszcze wyższe. Ceny mogą się różnić także w zależności od regionu oraz dodatkowych usług, takich jak szybka analiza wyników czy konsultacje z genetykiem.
Kiedy można zrobić test na ojcostwo?
Test na ojcostwo można wykonać w dowolnym momencie, gdy tylko dostępne są próbki od ojca i dziecka. Najczęściej testy na ojcostwo są wykonywane po narodzinach dziecka. Wówczas pobiera się próbki materiału od ojca i dziecka, zazwyczaj za pomocą wymazów z wnętrza policzka.
Czy można zrobić test DNA na ojcostwo przed narodzinami dziecka?
Istnieje możliwość wykonania testu na ojcostwo przed narodzinami dziecka, korzystając z metod nieinwazyjnych. Najczęściej stosowaną metodą jest analiza tzw. wolnego krążącego DNA płodowego obecnego w krwi matki. Badanie może być wykonane od około 9. tygodnia ciąży. Choć jest droższe i bardziej skomplikowane, oferują możliwość ustalenia ojcostwa bez ryzyka dla dziecka i matki. Inne metody, takie jak biopsja kosmówki czy amniopunkcja w celu pobrania próbki dziecka, są inwazyjne i wiążą się z pewnym ryzykiem dla płodu.
Czy testy na ojcostwo są wiarygodne?
Testy na ojcostwo są bardzo wiarygodne, szczególnie w przypadku dzisiejszego rozwoju technologii i nowoczesnych metod analizy DNA. Technologie takie jak sekwencjonowanie tysięcy polimorfizmów pojedynczego nukleotydu – SNP (Single Nucleotide Polymorphism), zapewniają wysoką precyzję wyników. Testy te mogą osiągnąć wiarygodność przekraczającą 99,99%, co oznacza, że ryzyko błędnych wyników jest bardzo niskie, a także pozwala na wykrywanie subtelnych różnic w DNA, które mogą być kluczowe w określaniu pokrewieństwa. Warto jednak wybierać laboratoria z dobrą reputacją i stosujące najnowsze technologie, aby zapewnić sobie pewność wyników.
Testy na ojcostwo są skutecznym narzędziem w ustalaniu biologicznego pokrewieństwa z szerokim zastosowaniem w różnych sytuacjach życiowych. Dzięki nowoczesnym metodom analizy DNA, wyniki są bardzo wiarygodne. Wybór odpowiedniego laboratorium oraz dostępnych metod jest kluczowy dla uzyskania dokładnych wyników. Testy te mogą być przeprowadzane zarówno po narodzinach dziecka, jak i w czasie ciąży, co pozwala na elastyczne podejście w różnych sytuacjach życiowych.
Bibliografia
1. Mayr WR., Brinkmann B., Rand S. Paternity testing–quo vadis? Blood Rev. 1991 Mar;5(1):51-4.
2. Zhang S., Han S., Zhang M., Wang Y. Non-invasive prenatal paternity testing using cell-free fetal DNA from maternal plasma: DNA isolation and genetic marker studies. Leg Med (Tokyo). 2018 May;32:98-103.
3. Tam JCW., Chan YM., Tsang SY et al. Noninvasive prenatal paternity testing by means of SNP-based targeted sequencing. Prenat Diagn. 2020 Mar;40(4):497-506.

















 – objawy, przyczyny, badania i leczenie")



 – działanie, nadmiar, niedobór i badanie poziomu hormonu wzrostu u dzieci i dorosłych")