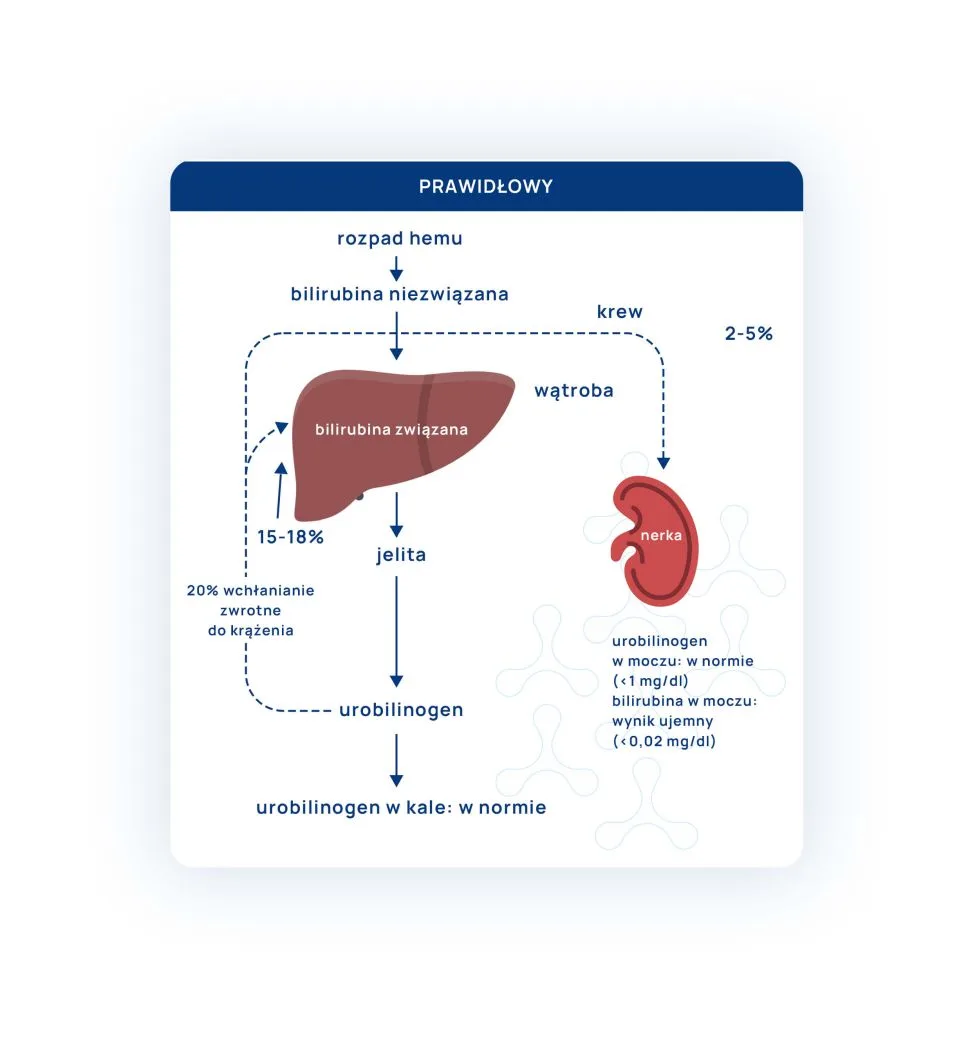
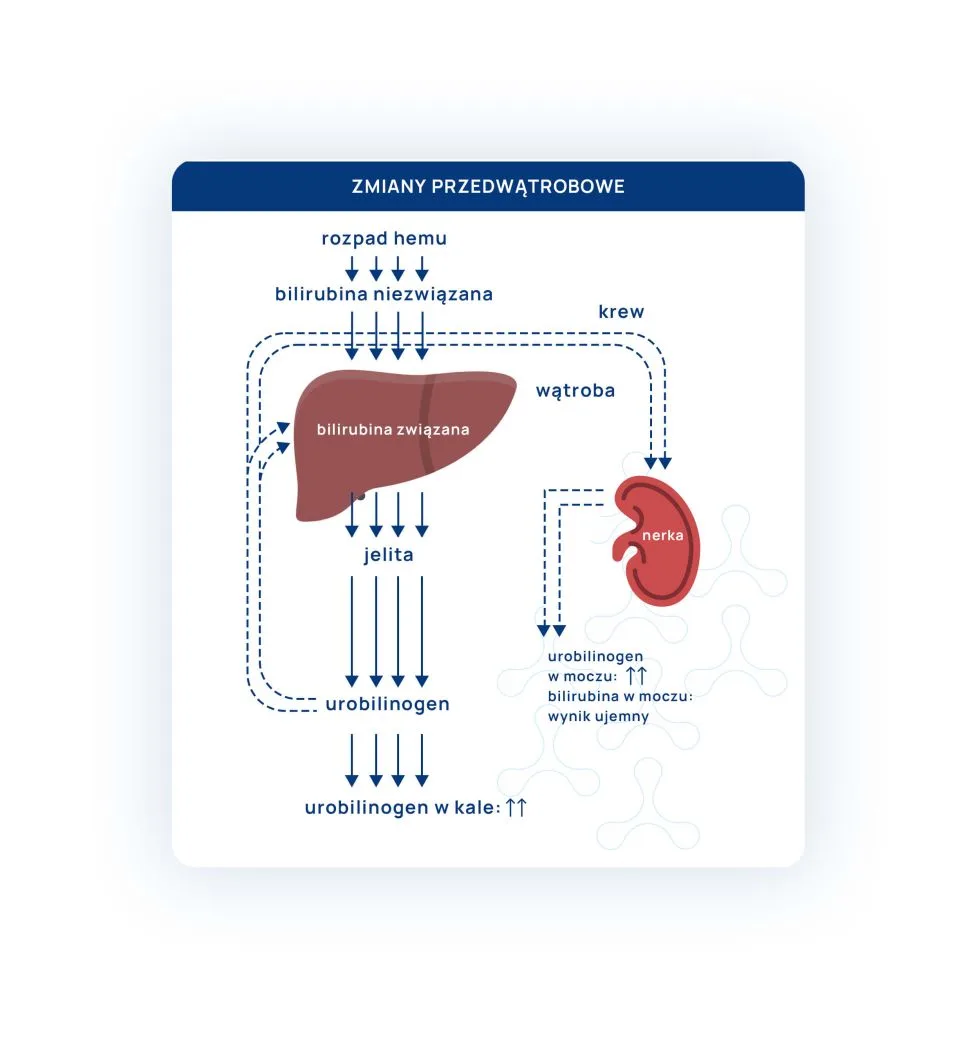
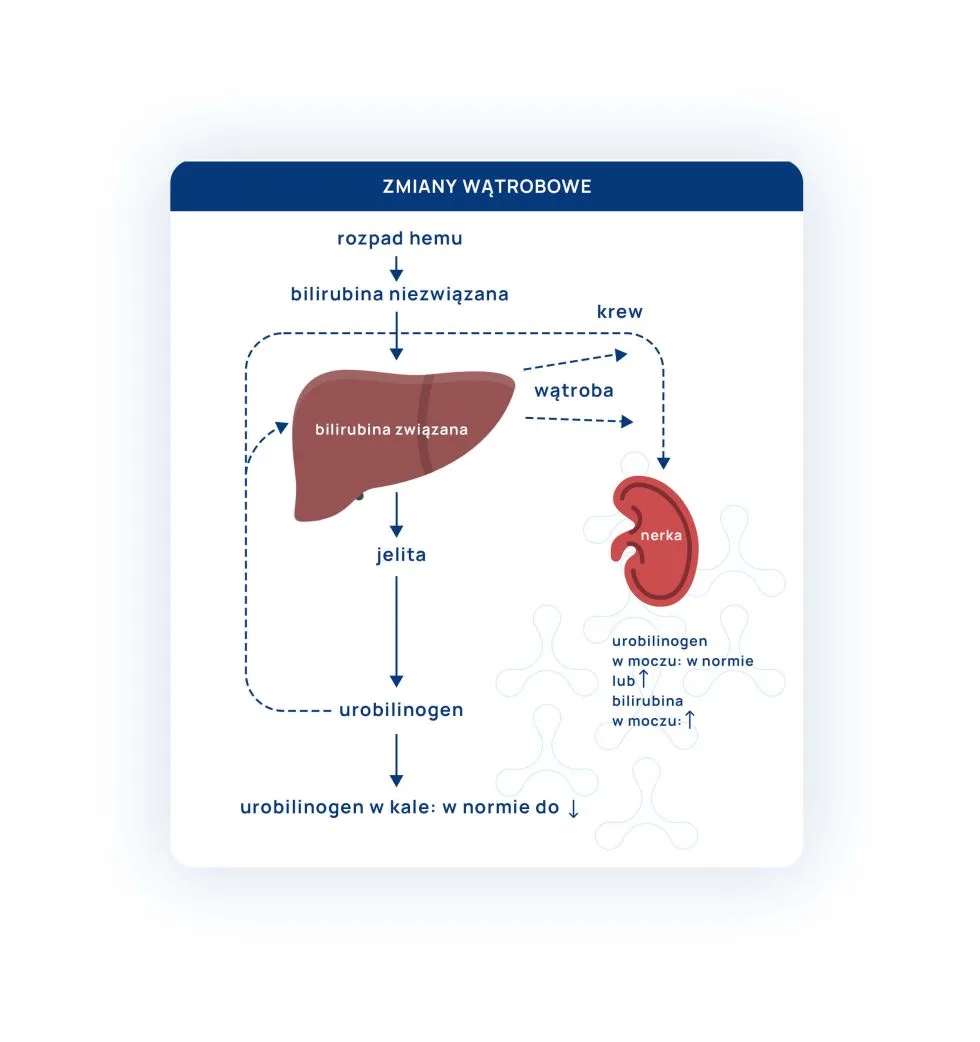
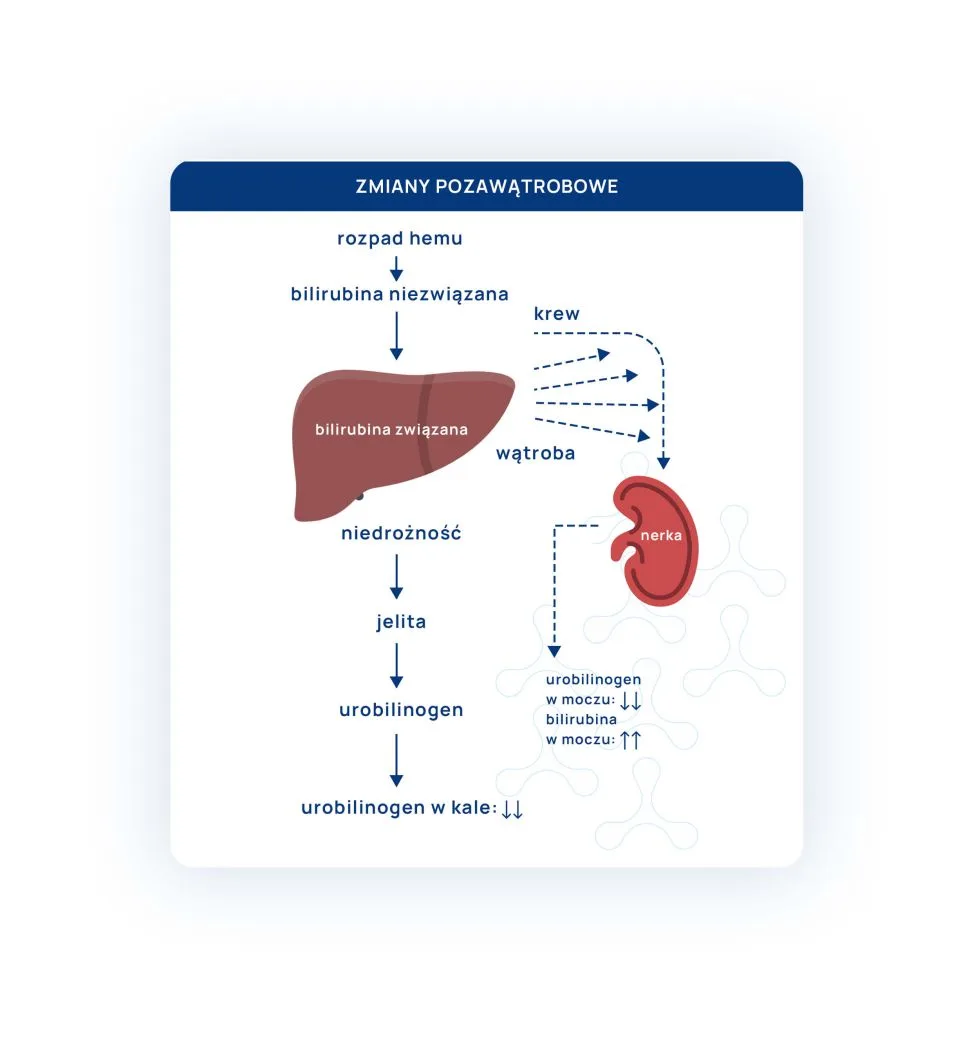
Poniższy artykuł jest zapisem webinaru, który odbył się 1.03.2023 r.
Reumatoidalne zapalenie stawów, znane również jako RZS, to przewlekła, systemowa choroba autoimmunologiczna, która atakuje przede wszystkim stawy, ale może wpływać również na wiele innych układów w ciele. Chociaż RZS jest często kojarzony z osobami starszymi, w rzeczywistości dotyka ludzi w każdym wieku, w tym tych w średnim wieku, aktywnych zawodowo.
RZS jest najczęstszą chorobą reumatyczną, często mylnie określaną jako reumatyzm. W skali globalnej szacuje się, że RZS dotyka około 1% populacji. W Polsce także mamy do czynienia z podobnym odsetkiem, co przekłada się na kilkaset tysięcy osób.
Jednym z często powtarzanych mitów na temat RZS jest przekonanie, że choroba ta obejmuje tylko stawy. W rzeczywistości RZS jest chorobą systemową, która może wpływać na wiele różnych układów w ciele, nie tylko na stawy.
Czy RZS to choroba osób starszych?
Innym często powtarzanym mitem jest wspomniane wcześniej przekonanie, że RZS jest chorobą starszych osób. Chociaż ryzyko wystąpienia RZS rzeczywiście zwiększa się z wiekiem, wiele osób rozwija chorobę w średnim wieku. Dlatego tak ważne jest szerzenie wiedzy na temat tej choroby i jej skutecznego leczenia, by pozwolić na utrzymanie jakości życia pacjentów na wysokim poziomie.
Ciąża a RZS
Częstym pytaniem dotyczącym RZS jest kwestia wpływu choroby na ciążę. W większości przypadków ciąża może przynieść ulgę w objawach RZS, choć choroba często powraca po porodzie. Trzeba jednak podkreślić, że RZS nie wpływa negatywnie na przebieg ciąży ani na zdrowie płodu. Zawsze jednak kobieta z RZS planująca ciążę powinna skonsultować swój stan zdrowia z lekarzem.
Objawy RZS
Reumatoidalne zapalenie stawów to przewlekła choroba, której cechą charakterystyczną jest zapalenie stawów. Jako choroba systemowa może wpływać na wiele różnych części ciała, nie tylko na stawy. Może dotykać m.in. narządu wzroku, układu sercowo-naczyniowego, układu oddechowego, czy układu nerwowego. Najczęstsze objawy RZS to:
- Ból i obrzęk stawów, zwłaszcza stawów dłoni i stóp, stawów śródręczno-paliczkowych i międzypaliczkowych bliższych oraz śródstopno-paliczkowych. Wiele osób zauważa symetryczne zajęcie stawów, co oznacza, że jeśli jeden staw jest dotknięty, staw po drugiej stronie ciała również. Nie jest to jednak regułą.
- Długa sztywność poranna – osoby z RZS często doświadczają sztywności stawów, zwłaszcza rano po przebudzeniu lub po okresie bezruchu. Taka sztywność może trwać godzinę lub dłużej.
- Utrata funkcji stawów – w miarę postępu choroby, stawy mogą stać się mniej elastyczne i mieć ograniczoną ruchomość. Może to utrudniać codzienne czynności, takie jak czesanie włosów, mycie zębów czy przygotowanie posiłków.
- Ogólne zmęczenie i brak energii
- Niewielka gorączka
- Utrata masy ciała
- Nocne poty
- Przewlekłe zmęczenie
- Problemy skórne, z sercem, płucami, oczami i naczyniami krwionośnymi
Objawy i powikłania pozastawowe RZS
Reumatoidalne zapalenie stawów (RZS) może wpływać na wiele układów w organizmie, w tym na układ oddechowy, krążenia, nerki czy układ okulistyczny. Przykładowo, RZS może prowadzić do zapalenia opłucnej, choroby śródmiąższowej płuc, guzków reumatycznych w sercu, kłębuszkowego zapalenia nerek czy zapalenia różnych warstw oka. Czasem RZS może wywoływać objawy, które mogą być mylące, jak kaszel, zaburzenia rytmu serca czy białkomocz. Te objawy mogą wskazywać na inne schorzenia, a ich przyczyną jest RZS. Mimo że jest to rzadkie, istnieje możliwość, że objawy pozastawowe mogą być bardziej zauważalne niż objawy stawowe, szczególnie gdy leczenie stawów jest skuteczne. Ważne jest, aby pamiętać, że leczenie RZS jest złożone i wymaga koordynacji między różnymi specjalistami.
Jak diagnozujemy RZS?
Diagnozowanie reumatoidalnego zapalenia stawów (RZS) to proces, który obejmuje kilka ważnych kroków:
- Wywiad: to podstawowe narzędzie diagnostyczne w reumatologii, stanowiące około 85-90% sukcesu. Lekarz rozmawia z pacjentem, starając się zdobyć jak najwięcej informacji. Ważne są pytania o objawy, takie jak: które stawy są zajęte, czy występuje obrzęk, czy występuje sztywność poranna, która może wskazywać na chorobę zapalną. Co więcej, pytania dotyczące historii medycznej pacjenta i jego rodziny są istotne, ponieważ niektóre choroby mogą prowadzić do dolegliwości stawowych, a genetyka może wpływać na predyspozycje do RZS.
- Badanie fizykalne: w kolejnym kroku lekarz dokonuje badania przedmiotowego, które obejmuje ocenę stawów pacjenta. Sprawdza ruchomość stawów, czy występuje obrzęk i czy stawy są bolesne. Obrzęk może być delikatny, ale wyczuwalny pod palcami, co wskazuje na zapalenie.
- Wywiad rodzinny: pytania dotyczące chorób reumatycznych w rodzinie pacjenta są również istotne. Jeśli bliscy krewni (rodzice, dzieci, rodzeństwo) cierpieli na RZS, zwiększa to ryzyko wystąpienia tej choroby u pacjenta.
Trzeba jednak pamiętać, że brak historii choroby w rodzinie nie wyklucza możliwości zachorowania na RZS. Podobnie, obecność symptomów, takich jak obrzęk stawów czy sztywność poranna, nie oznacza automatycznego diagnozowania RZS. Dlatego diagnostyka tej choroby zawsze wymaga kompleksowego podejścia i analizy wszystkich dostępnych danych.
Diagnostyka laboratoryjna w wykrywaniu RZS
Reumatoidalne zapalenie stawów (RZS) to przewlekła choroba autoimmunologiczna, która prowadzi do zapalenia i zniszczenia stawów. Diagnostyka laboratoryjna odgrywa kluczową rolę w jej wykrywaniu, zapewniając ścisłe i szybkie wykrywanie oraz umożliwiając monitorowanie postępów choroby. Podstawowe badania laboratoryjne, które są częścią diagnostyki RZS, obejmują badania krwi na obecność specyficznych przeciwciał: reumatoidalnego czynnika RF oraz przeciwciał przeciwko cyklicznemu peptydowi cytrulinowanemu (anty-CCP).

Choć badanie RF i anty-CCP jest często wykorzystywane w diagnostyce RZS, nie jest ono specyficzne tylko dla tego schorzenia. Obecność tych przeciwciał może być wykryta również w przypadku innych chorób autoimmunologicznych. Dlatego, aby zwiększyć precyzję diagnostyczną, lekarze często korzystają z dodatkowych badań, takich jak pomiar stężenia białka C-reaktywnego (CRP) i szybkości sedymentacji erytrocytów (OB), które wskazują na ogólny stan zapalny organizmu.
Rozpoznanie RZS jest procesem stopniowym i wieloaspektowym, uwzględniającym zarówno wyniki badań laboratoryjnych, jak i obraz kliniczny pacjenta. Diagnostyka laboratoryjna, mimo że nie jest jedynym elementem w diagnostyce RZS, stanowi istotne narzędzie, pozwalające na szybkie rozpoznanie choroby, monitorowanie jej przebiegu oraz efektywność stosowanego leczenia. Szczególnie ważna jest dla osób z rodzinną historią chorób autoimmunologicznych, u których ryzyko wystąpienia RZS jest wyższe.
Leczenie reumatoidalnego zapalenia stawów
Leczenie reumatoidalnego zapalenia stawów (RZS) jest skomplikowane i wielopłaszczyznowe, zaczynając od edukacji pacjenta o chorobie, jej przebiegu i potencjalnych powikłaniach, po wsparcie psychologiczne i psychiatryczne w radzeniu sobie z przewlekłym bólem i obniżonym nastrojem, które mogą prowadzić do depresji.
Ważnym elementem terapii są leki. Lekiem pierwszego rzutu jest metotreksat, który jest skuteczny, ale efekty jego działania mogą pojawić się dopiero po 6-8 tygodniach. Inne dostępne leki to m.in. leflunomid, sulfasalazyna oraz chlorochina i hydroksychlorochina. W przypadku niskiej aktywności choroby, Amerykańskie Stowarzyszenie Reumatologiczne zaleca stosowanie hydroksychlorochiny.
Kiedy te leki nie przynoszą oczekiwanych efektów, stosuje się leki syntetyczne, celowane, które wpływają na układ zapalny. To inhibitory kinazy janusowej (np. Barycytynib, Upadacytynib i Tofacytynib) – leki innowacyjne, które zmniejszają stan zapalny.
Jeszcze inną grupą leków są leki biologiczne i biopodobne, które działają przyczynowo na cząsteczki antyTNF i TNF, czynniki zwiększające stany zapalne. Przykłady takich leków to Adalimumab, Certolizumab, Etanercept.
Oprócz powyższych, w przypadku silnego bólu, stosowane są również niesteroidowe leki przeciwzapalne oraz, na krótko, sterydy.
Inne możliwości leczenia – fizjoterapia, reumatologia, wsparcie psychologiczne
Oprócz farmakoterapii, reumatoidalne zapalenie stawów można leczyć za pomocą wielu innych metod. Fizjoterapia, która obejmuje kinezyterapię, skupia się na zwiększeniu siły mięśniowej, poprawie ruchomości stawów oraz zmniejszeniu ryzyka wystąpienia niepełnosprawności. Następnie mamy fizykoterapię, której głównym celem jest zmniejszenie dolegliwości bólowych poprzez stosowanie termoterapii, balneoterapii, laseroterapii i elektroterapii. Te techniki, stosowane indywidualnie lub łącznie, przyczyniają się do złagodzenia objawów choroby.
W sytuacjach, gdy stawy pacjenta uległy destrukcji i inne metody leczenia nie przynoszą oczekiwanych efektów, możliwe jest zastosowanie interwencji chirurgicznej w ramach reumoortopedii. Ortopedzi mogą usztywnić staw lub dokonać jego wymiany, co znacznie poprawia jakość życia pacjenta.
Ponadto, niewątpliwie ważnym aspektem procesu leczenia jest wsparcie psychologiczne. Życie z przewlekłym bólem i świadomością ciężkiej choroby autoimmunologicznej może prowadzić do obniżenia nastroju i potencjalnie do poważnych zaburzeń psychiatrycznych. Wsparcie ze strony psychologów lub psychiatrów jest niezbędne w procesie leczenia, pomagając pacjentom normalnie funkcjonować mimo choroby i zapobiegając ryzyku zgonu związanego z ciężkimi zaburzeniami psychicznymi.
Czy RZS można wyleczyć?
RZS, czyli reumatoidalne zapalenie stawów, jest chorobą przewlekłą, a więc niestety nie da się jej wyleczyć. Mit, że RZS można wyleczyć, musimy obalić. Prawdziwym celem w leczeniu RZS jest osiągnięcie remisji lub utrzymanie niskiej aktywności choroby.
Remisja to stan, w którym mimo obecności choroby, nie odczuwamy jej objawów. To trochę jak z nadciśnieniem tętniczym: mimo że mamy diagnozę, stosujemy leki, które regulują nasze ciśnienie i nie odczuwamy objawów. Tak samo jest z RZS – stosujemy leki, nie mamy objawów, ale gdybyśmy je odstawili, objawy najprawdopodobniej wróciłyby. Remisja to osiągnięcie stanu, w którym choroba jest „wyciszona”, ale nie zniknęła.
Aby osiągnąć remisję, kluczowe jest realizowanie zaleceń lekarskich. Musimy stosować odpowiednie leki, w odpowiednich dawkach i formie oraz robić to systematycznie. Ważne jest również utrzymanie dobrej relacji z lekarzem, zaufanie mu i słuchanie jego zaleceń.
RZS to nie wyrok
Mimo że diagnoza RZS może być szokiem dla pacjenta, nie jest to koniec świata. Rozwój medycyny pozwolił na skuteczne leczenie tej choroby, które może doprowadzić do remisji, pozwalając pacjentom żyć normalnym życiem. Ważne jest jednak szybkie wykrycie choroby i przestrzeganie zaleceń lekarskich, w tym stosowanie przepisanych leków zgodnie z zaleceniami.